Oxygen Evolution on Single Cobalt Sites with an Intramolecular Hydroxyl Nucleophilic Attack Pathway
Published in Chemistry

Oxygen evolution reaction (OER) is an ideal anodic reaction that can provide electrons and protons for hydrogen generation, CO2 and N2 reduction, and other electrochemical reactions. However, OER is kinetically slow because of the complicated removal of four protons and electrons from two water molecules, O-H bonds cleavage, and attendant O-O bond formation. Thus, the design of efficient, stable electrocatalysts for OER is needed; and an in-depth understanding of the OER mechanism and structure-function relationship of catalysts is indispensable for the development of advanced OER systems.
Several strategies, such as morphology control, component and defect engineering, have been developed in recent years to improve the activity of heterogeneous catalysts for the OER; however, deep understandings of the mechanism for heterogeneous catalysts at the molecular level are still rarely reported. If we re-think the OER itself, the mass of the proton is much heavier than that of the electron; meanwhile, the O−H bond cleavage involves an endothermic process (the bond dissociation energy of the H−OH bond is as high as 118 kcal mol–1). Therefore, the activation of O−H bond associated with the O−O bond formation is a particular challenge. Based on this insight, the effective activation of the O−H bond of water and the acceleration of the interfacial equivalent proton transfer of the rate-determining step (RDS) is considered as a worthy direction to be devoted to the design of more efficient OER catalysts.
Our recently comprehensive mechanism studies (Nat. Commun., 2019, 10, 5074; Sci. China Chem., 2022, 65, 382-390) have revealed that if the RDS of the OER involves proton transfer, installing proton acceptors such as carboxylate and sulfonate groups in the out coordination sphere of the nearby the catalytic centers could serve as relays to accelerate the rate of proton transfer and water oxidation. These discoveries suggested that promoting the interfacial proton transfer of the RDS for OER could improve the activity of a catalyst. Then, an obvious question was raised: if the interfacial proton transfer process no longer controls the reaction rate, could the catalyst obtain a faster reaction rate?
To answer this question, our research group and collaborators report a molecularly well-defined heterogeneous OER catalyst with Aza-fused-π-conjugated-microporous-polymer (Aza-CMP) coordinated with single cobalt sites (Aza-CMP-Co). The molecular nature of the isolated catalytic sites makes Aza-CMP-Co a reliable model for the study of heterogeneous water oxidation mechanism. The single cobalt sites in Aza-CMP-Co exhibited superior activities to those of most cobalt-based electrocatalysts under alkaline and near-neutral conditions. More importantly, kinetics data shows that the intrinsic catalytic OER activity of the cobalt sites in Aza-CMP-Co under alkaline conditions is a hundred times faster than that under near-neutral conditions at the same overpotentials. As for a typical heterogeneous catalytic reaction, the reaction rate of the active sites should only depend on the catalytic driving forces but not on the concentration of reactant. This unusual phenomenon caught our attention, and comprehensive mechanism studies were conducted.
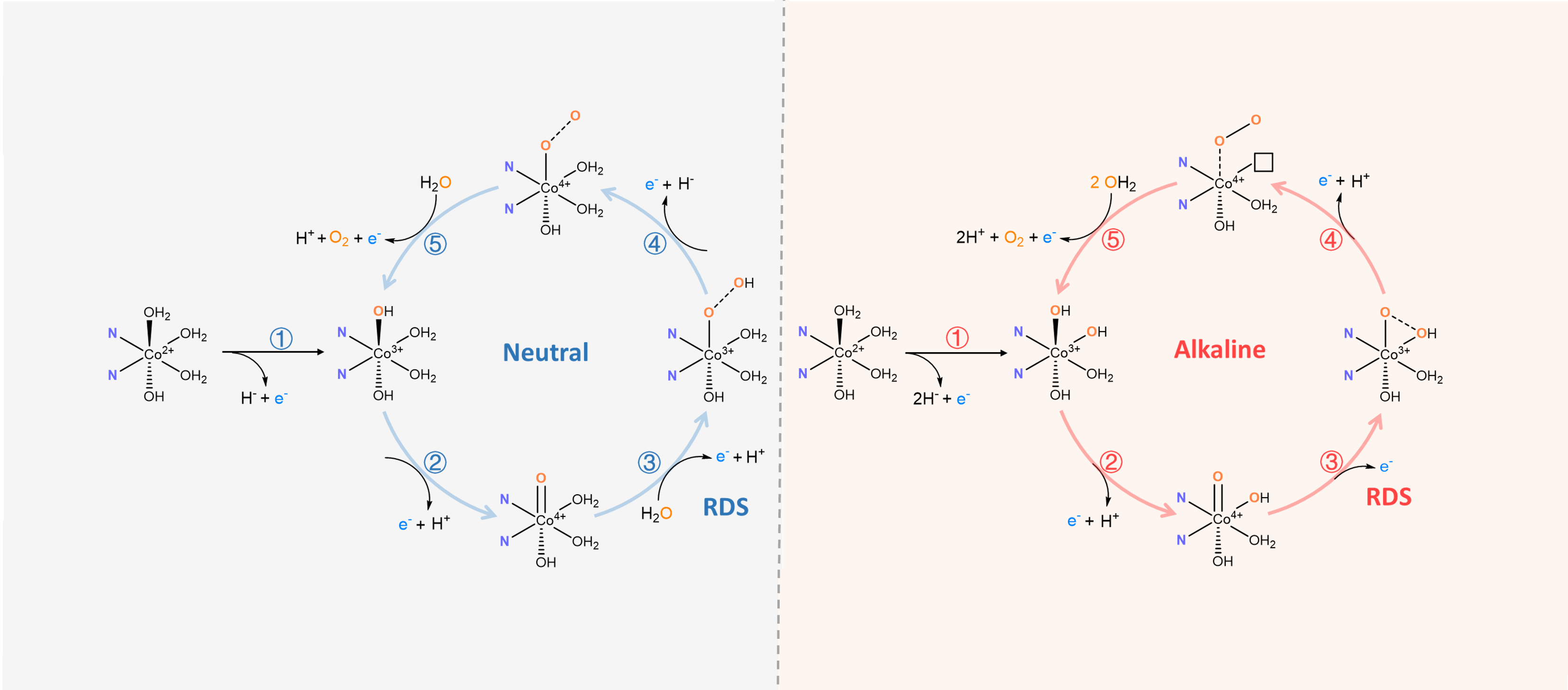
.png "Fig. 1")
Fig. 1 Proposed OER mechanisms. concerted PCET processes trigger the intermolecular WNA pathway under neutral conditions, and the the non-concerted PCET process in Co2+/3+ redox facilitates the IHNA pathway under the basic pH range.
Pourbaix diagram, deuterium kinetic isotope effects, quantitative cyclic voltammetry analysis, proton inventory, anion and cation effects results revealed that O−O bond formation pathways (the RDS) of the single cobalt sites in Aza-CMP-Co are different under alkaline and neutral conditions. And this transition of the O−O bond formation pathways was controlled by the pKa of the Co3+ intermediates, acid dissociation, and PCET features of the pre-redox states. When the pH of the electrolyte is larger than the pKa value of the Co3+ intermediates, electrochemically driven deprotonation results in the transport of an additional proton, as shown in Fig. 1, which facilitates the intramolecular hydroxyl nucleophilic attack (IHNA) pathway where the adjacent OH− attacks Co4+=O to form the O-O bond. However, when the pH of the electrolyte is smaller than the pKa value of Co3+, step-by-step concerted PCET processes will occur with an intermolecular water nucleophilic attack (WNA) pathway as the RDS for the O−O bond formation. Compared to the intermolecular WNA pathway, diffusion-controlled interfacial proton transfer and endothermic O−H bond cleavage processes are not involved in this IHNA pathway, which leads to faster reaction kinetics. The activation energies established through temperature-controlled experiments and DFT calculations also show that the intramolecular IHNA pathway requires much lower activation energy compared with that of the intermolecular WNA pathway.
We believe that this study may provide significant insights into the crucial function of the pH for the electrolyte and enhancement of the OER activity of catalysts by regulating the IHNA pathway.
For detailed information, please see our article " Intramolecular Hydroxyl Nucleophilic Attack Pathway by a Polymeric Water Oxidation Catalyst with Single Cobalt Sites" in Nature Catalysis 5, 414–429 (2022)
Fusheng Li is working on Physical Chemistry. His main research goal is to develop catalysts and devices for solar-chemical transformations of relevance to synthesis, energy and sustainability. Current research topics include: (i) reaction mechanisms of artificial catalysts for water splitting; (ii) photoelectrochemical (PEC) cells for solar fuels production; (iii) proton-coupled electron transfer (PCET) reactions.
Follow the Topic
-
Nature Catalysis
This journal brings together researchers from across all chemistry and related fields, publishing work on homogeneous catalysis, heterogeneous catalysis, and biocatalysts, incorporating both fundamental and applied studies.




Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in