Phenogenomic analyses - linking bacterial genotypes and function
Published in Microbiology

Genome-wide genetic screens are unparalleled tools linking genes to bacterial phenotypes. Its simplicity, demonstrating that a mutant causes a change in bacterial behaviour, is its strength but also its weakness. Biology is complex with an almost endless number of genetic backgrounds, gain- and loss-of-function variations and combinations thereof, conferring single or multiple phenotypes. This genetic and phenotypic diversity of many bacterial species that have naturally evolved over decades is now more easily accessible. We can characterise thousands of bacterial isolates via whole genome sequencing and phenotypic profiling. Similar to forward genetic screens, genotypes can be linked to phenotypes to reveal molecular networks underlying bacterial function. While this step is relatively simple in mutant screens, it remains the bottleneck of genome-wide association studies (GWAS), which iteratively assess the association of each genotype with a particular phenotype. Due to epistasis, linkage disequilibrium, population structure and other constraints the approach’s utility is limited and often only a small fraction of the underlying biology is captured; in other words, the vast genetic diversity of naturally occurring samples may be too complex to understand. Within a collaboration of scientists with very different expertise and led by Andres Floto (University of Cambridge, UK), we aimed to advance the current concept of GWAS into a multi-modular strategy, named phenogenomic analyses, to capture the complexity of genotype-phenotype associations.
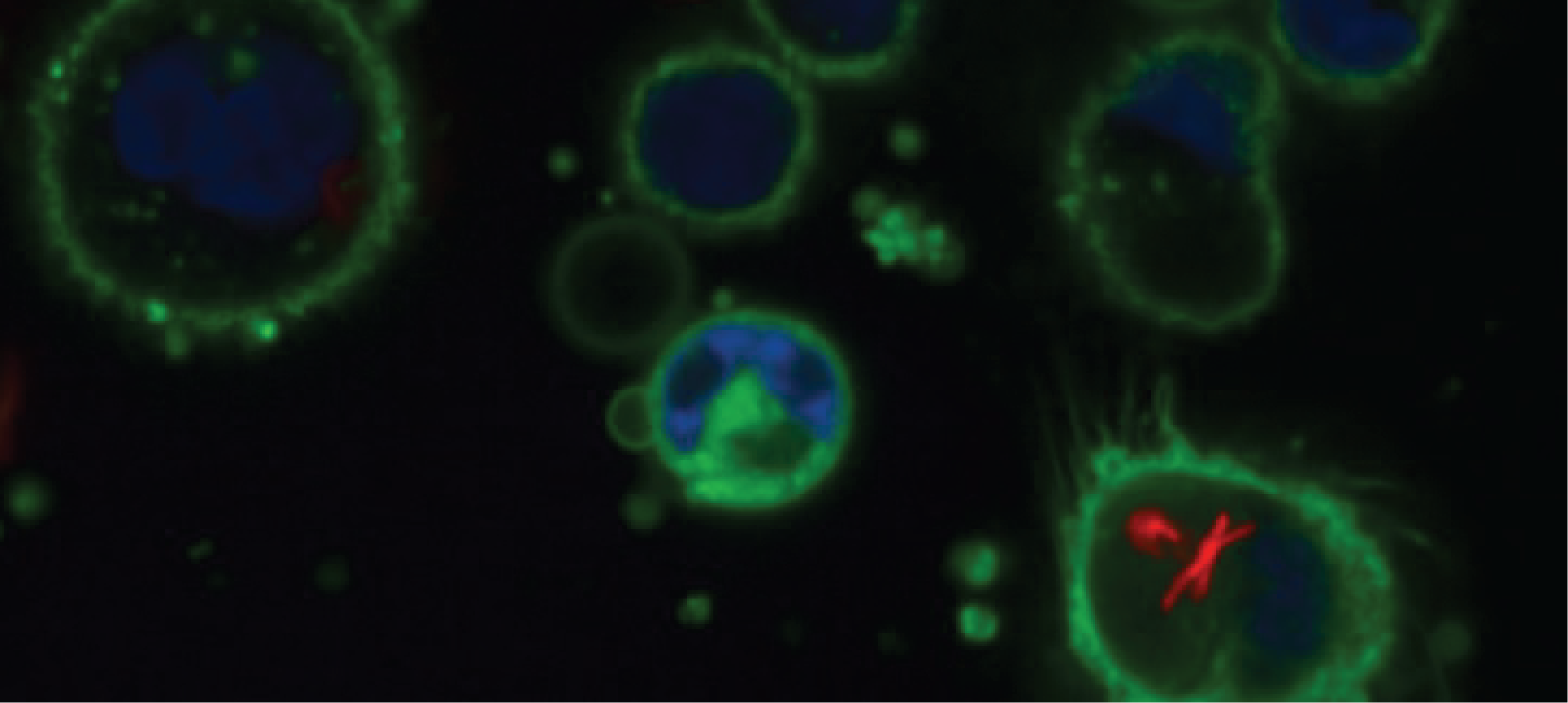
We employed our strategy on Mycobacterium abscessus, a particularly difficult-to-treat but poorly understood pathogen that causes increasing numbers of respiratory infections globally. Our first goal was to phenotypically profile several hundred clinical isolates from respiratory samples of patients with M. abscessus lung disease. We aimed for in vitro and in vivo phenotypes, reflecting bacterial infection and therapy. We used serial OD measurements across different growth conditions and standard MIC testing to assess basic bacterial and drug response characteristics. We then expanded to the next level of mycobacterial infection, namely macrophage infection. Rather than applying standard macrophage infection assays, we established a high-content screen for THP-1 cells. After several months of work and many unsuccessful attempts to reliably stain M. abscessus, we almost stopped this strategy. Only by transforming M. abscessus with a fluorophore (and therefore several hundred clinical isolates), we obtained the necessary mycobacterial signal for imaging. Automatic image acquisition and analysis of thousands of single cells and bacteria was much less time-consuming and provided a very high resolution for downstream analysis. Lastly, we wanted to integrate in vivo data for assessing bacterial virulence and potentially host responses into our multi-dimensional phenotyping approach. However, only a few models exist that allow, due to technical, financial and ethical considerations assessing thousands of animals. So, we teamed up with Marc Dionne’s group (Imperial College London, UK) to explore the Drosophila infection model [1, 2] for M. abscessus and eventually infected with the help of Will Pearson, more than 10.000 flies to evaluate M. abscessus virulence and the Drosophila host response of each isolate. Using these experimental phenotypic dimensions of M. abscessus and pairing them with clinical data, we identified isolate clusters that had particular phenotypes in common and were associated with patient outcomes.
We next aimed to reveal the molecular networks underlying these bacterial behaviours. To expand traditional bioinformatical strategies and integrate different know how we collaborated with the groups of Julian Parkhill and Tom Blundell (both University of Cambridge, UK). In a first step, we performed conventional genome-wide association studies across all phenotypes. A significant limitation of GWAS is linkage disequilibrium, the non-random association of genetic variants due to clonal progeny and infrequent recombination events. By applying a graph-based approach [3] coupled with proteome-wide in silico modelling of M. abscessus [4], both developed in Tom Blundell’s team, we predicted the putative effects of missense mutations, thereby selecting the most deleterious variants and overcoming some constraints of LD. Another limitation is the ignorance of epistasis, i. e. the interaction of genotypes for conferring a phenotype. Marcin Skwark has established direct coupling analysis (DCA) to study genomic coevolution [5] and has now applied and integrated a more efficient DCA strategy [6], considering around 1012 possible couplings. With this strategy, we could identify many coevolving and likely interacting genes on a genome-wide scale. Finally, to validate many of these findings and tools, we used the M. abscessus CRISPR interference platform Sophie Burbaud developed and could replicate some of our GWAS, mCSM and DCA findings including two so far unknown virulence factors of M. abscessus infection.
In summary, we aimed to advance genotype-phenotype inference from a simple association (GWAS) to a multi-modal concept of assessing molecular networks underlying phenotypic diversity. Though, phenogenomic analyses shouldn’t be considered a complete tool but rather a strategy that we and hopefully others will continue to improve, to get a better molecular understanding of bacterial behaviours.
REFERENCES
- Dionne MS, Ghori N, Schneider DS (2003) Drosophila melanogaster is a genetically tractable model host for Mycobacterium marinum. Infection and immunity 71:3540–3550. https://doi.org/10.1128/iai.71.6.3540-3550.2003
- Pean CB, Schiebler M, Tan SWS, et al (2017) Regulation of phagocyte triglyceride by a STAT-ATG2 pathway controls mycobacterial infection. Nature Communications 8:1–11. https://doi.org/10.1038/ncomms14642
- Pires DEV, Ascher DB, Blundell TL (2014) mCSM: predicting the effects of mutations in proteins using graph-based signatures. Bioinformatics 30:335–342. https://doi.org/10.1093/bioinformatics/btt691
- Skwark MJ, Torres PHM, Copoiu L, et al (2019) Mabellini: a genome-wide database for understanding the structural proteome and evaluating prospective antimicrobial targets of the emerging pathogen Mycobacterium abscessus. Database : the journal of biological databases and curation 2019:4471. https://doi.org/10.1093/database/baz113
- Skwark MJ, Croucher NJ, Puranen S, et al (2017) Interacting networks of resistance, virulence and core machinery genes identified by genome-wide epistasis analysis. PLoS genetics 13:e1006508. https://doi.org/10.1371/journal.pgen.1006508
- Gao C-Y, Cecconi F, Vulpiani A, et al (2019) DCA for genome-wide epistasis analysis: the statistical genetics perspective. Physical biology 16:026002. https://doi.org/10.1088/1478-3975/aafbe0
Follow the Topic
-
Nature Microbiology
An online-only monthly journal interested in all aspects of microorganisms, be it their evolution, physiology and cell biology; their interactions with each other, with a host or with an environment; or their societal significance.





Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in