Recessive ReNU2 syndrome: highly prevalent and potentially treatable
Published in Neuroscience, Protocols & Methods, and Cell & Molecular Biology
Over more than a decade, my research group has developed computational tools and statistical methods for discovering the etiologies of rare diseases using whole-genome sequencing (WGS) and phenotype data from large patient collections. We first applied our methods (Greene et al., American Journal of Human Genetics 2017; Turro et al., Nature 2020) to data generated by the NIHR BioResource (NBR)–Rare Diseases at the University of Cambridge, the first major rare disease project to use WGS. In contrast to whole-exome sequencing (WES) technology, which only reads protein coding genes (approximately 2% of the genome), WGS reads the entire genome. The NBR project in Cambridge was a precursor to the 100,000 Genomes Project (100KGP) established by the UK government through its subsidiary, Genomics England Ltd, which now supports the NHS’s Genomic Medicine Service. Once the 100KGP collection was complete, we began applying the methods we had developed for the NBR project to the new larger dataset.
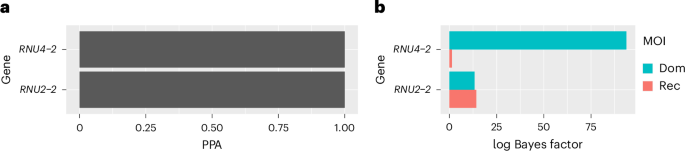
By June 2021, we had identified over 200 coding and noncoding genetic associations with 269 rare diseases. Most of the associations were already known. However, they included a very strong, previously unreported, dominant association between a small nuclear RNA (snRNA) gene called RNU4-2 and neurodevelopmental disorders (NDDs). We had also identified 19 compelling novel associations with coding genes, including previously unknown genetic causes of Loeys-Dietz syndrome, primary lymphedema and congenital deafness. Because we are a small team, we decided to pursue the coding associations first (Greene et al., Nature Medicine 2023) before returning to noncoding genes.
In May 2024, we published a paper describing the dominant RNU4-2 NDD (Greene et al., Nature Medicine 2024). This condition, now called ReNU syndrome, was identified independently from the same data by a separate group (Chen et al., Nature 2024). It is the most prevalent known autosomal dominant NDD. A minor refinement of our analysis allowed us to identify a second dominant association between NDDs and RNU2-2P, a gene that was then considered to be a pseudogene (as the P suffix indicates). Following this finding, the gene-naming body HGNC removed its pseudogene designation and renamed it RNU2-2 (Greene et al., Nature Genetics 2025). This disorder, now known as dominant ReNU2 syndrome, has a prevalence roughly a fifth of that of ReNU syndrome.
This brings us to our latest work (Greene et al, Nature Genetics 2026). Although we had detected a strong dominant association between RNU2-2 and NDDs, our Bayesian method also identified strong evidence of a recessive association. Thanks to the short length of the gene (191 bp), we were able to phase the variant alleles along the entire gene using a technique called read-backed phasing. We refined our genetic association methodology to take the phasing information into account, improving its ability to discriminate between disease-causing and benign variants. This allowed us to identify almost 50 biallelic patients with a credible diagnosis of a recessive form of ReNU2 syndrome. It is, by a wide margin, the most prevalent known recessive NDD. Including affected siblings within families, we estimate that it accounts for 60% as many cases as ReNU syndrome, and 10% of all recessive NDD cases with a known genetic etiology. The high prevalence of this novel recessive syndrome is particularly intriguing because the most common monogenic NDDs tend to be dominant rather than recessive. RNU2-2's unusually high mutation rate may explain the high prevalence.
RNU4-2 and RNU2-2 are transcribed respectively into U4-2 and U2-2 snRNAs, paralogs of well-known components of the major spliceosome. Our analysis of whole blood RNA sequencing data from patients and controls showed that recessive ReNU2 syndrome patients have substantially reduced levels of U2-2 snRNA in their cells, while patients with dominant ReNU/ReNU2 syndromes have normal levels of U4-2/U2-2 snRNA. Carriers of recessive RNU2-2 alleles, who are unaffected, have very low levels of mutant U2-2 snRNA, but their wild type U2-2 allele is upregulated. It appears that patients with higher (albeit still low) levels of U2-2 are less severely affected. This suggests that introducing even a small amount of exogenous wild type U2-2 RNA into the appropriate compartments of disease-mediating cells in patients (e.g., neurons) could be therapeutic. For example, it might reduce the incidence of seizures and ameliorate irregular development. Furthermore, the variance in expression among unaffected individuals is substantial, suggesting that toxicity due to dose overshooting would not be a safety concern.
Prior to the recent identification of RNU gene-mediated NDDs by us (Greene et al., Nature Medicine 2024; Greene et al., Nature Genetics 2025; Greene et al., Nature Genetics 2026) and other groups (Chen et al., Nature 2024; Jackson et al., Nature Genetics 2025; Nava et al., Nature Genetics 2025; Leitão et al., Nature Genetics 2026; Jackson et al., Nature Genetics 2026), none of the five sets of snRNA paralogs related to the major spliceosome had been implicated in disease. Using Genomic Medicine Service data to compare the number of patients with ReNU syndrome with the number of patients with Rett syndrome, which has a well-estimated global prevalence (of approximately 1 in 28,000 individuals of either sex), suggests there may be several hundred thousand individuals with ReNU or ReNU2 syndrome worldwide. Our findings will allow these patients, and those yet to be born, to receive a genetic diagnosis, provided sequencing of RNU4-2 and RNU2-2 is made widely available. To help issue such diagnoses and to learn more about these conditions, my colleagues and I have established the INDEED study, through which we are collecting natural history data and acquiring biospecimens, which we hope will lay a foundation for future trials.
Acknowledgements: thank you to my co-authors, particularly my long-term collaborator Daniel Greene. Our work would not have been possible without the data generated by Genomics England and the participation of large numbers of rare disease patients and their families.
Follow the Topic
-
Nature Genetics
This journal publishes the very highest quality research in genetics, encompassing genetic and functional genomic studies on human and plant traits and on other model organisms.

Related Collections
With Collections, you can get published faster and increase your visibility.
Mechanisms and impact of 3D genome organisation
Publishing Model: Hybrid
Deadline: Nov 30, 2026
Human Admixture and Migration
Publishing Model: Hybrid
Deadline: Feb 28, 2027
Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in