Recovery of nearly 8,000 metagenome-assembled genomes substantially expands the tree of life
Published in Microbiology
The paper in Nature Microbiology is here: http://go.nature.com/2w3O6xX
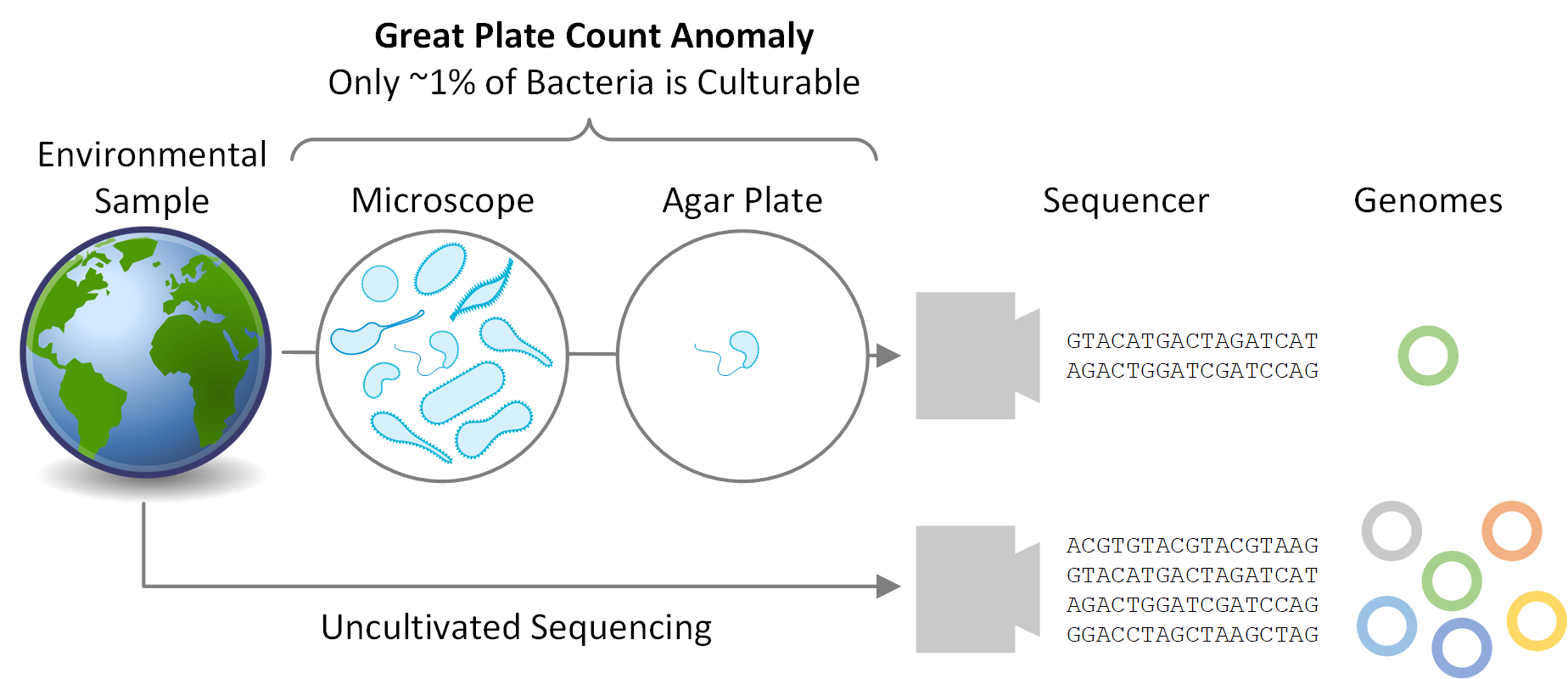
Much of microbial diversity remains to be discovered. Staley and Konopka coined the term “great plate count anomaly” in 1985 to describe the observation that the majority of microbes seen under a microscope are not amenable to being grown under laboratory conditions. The prevalence of bacterial and archaeal species that remain challenging, if not impossible, to cultivate is due to a variety of factors including slow growth rates, fastidious growth requirements, and strict dependencies on syntrophic relationships. This has hampered scientific investigations as cultivation has, until recently, been necessary to obtain sufficient DNA to reconstruct a microbe’s genome. However, this requirement has been removed in the last few years thanks to advances in sequencing technology and computational techniques that allow microbial genomes to be readily recovered directly from environmental samples, bypassing the need for laboratory cultivation.
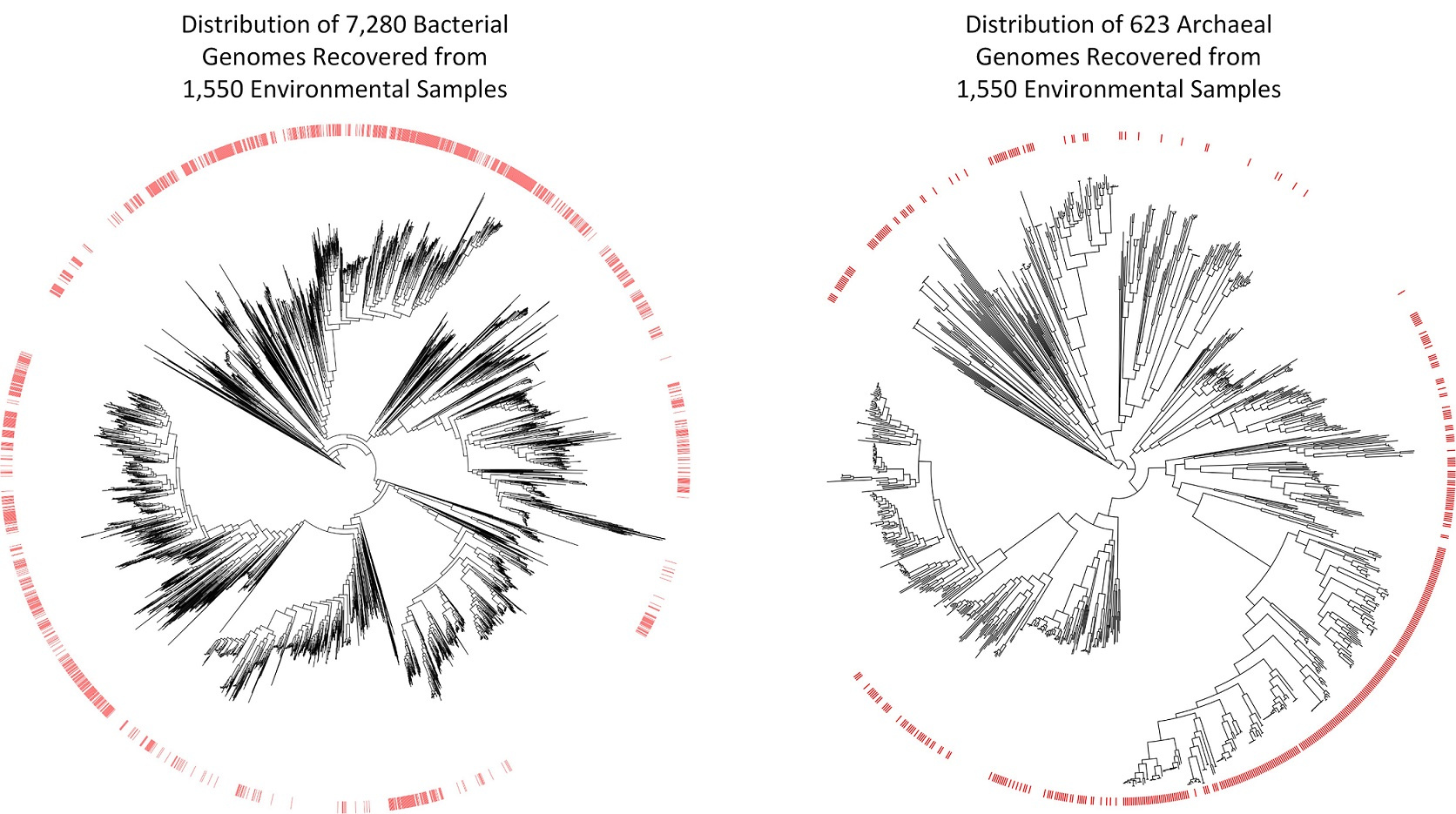
The scientific community has been collecting and sequencing environmental samples for nearly two decades and has been making this data available through sequencing repositories such as the Sequence Read Achieve (https://www.ncbi.nlm.nih.gov/sra). Realizing this data harbored the genetic information of numerous microbial species currently unknown to science, we decided to examine it with the aim of recovering novel microbial genomes. Since obtaining genomes from environmental samples requires substantial computational resources, we focused on 1,550 samples predominately of non-human origin as this allowed us to consider a range of environments containing a large diversity of microbial populations. We obtained 7,280 bacterial and 623 archaeal genomes from these environmental samples, nearly a 10% increase over the approximately 80,000 genomes currently in genome repositories. However, the real value of these genomes is that many of them are evolutionarily distinct from previously recovered genomes. They increase the evolutionary diversity spanned by both bacterial and archaeal genome trees by over 30%, and are the first representatives within 17 bacterial and three archaeal phyla.
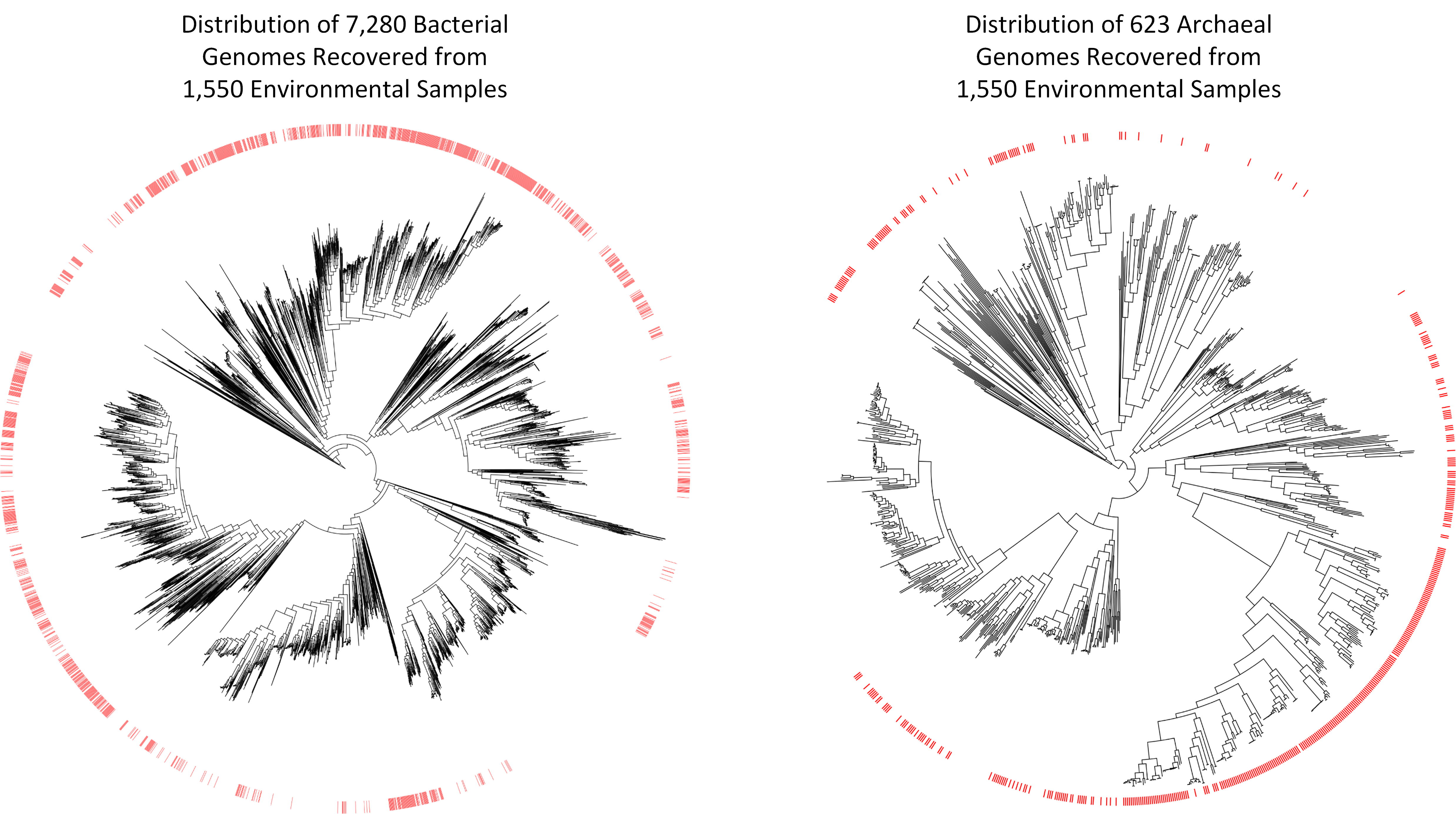
The approximately 8,000 genomes we have recovered move us closer to a comprehensive genomic representation of the microbial world, but also show that much remains to be discovered. However, for the first time we have the required tools to make substantial inroads into the vast diversity of microbial life. We anticipate that processing of environmental samples deposited in other public repositories such as the Integrated Microbial Genomes and Metagenomes (IMG/M) database and Metagenomics Rapid Annotation Server (MG-RAST) will add tens of thousands of additional microbial genomes to the tree of life, and numerous studies have been published during the completion of this research which have reported dozens or hundreds of evolutionarily diverse genomes from varying environments. The tools for obtaining genomes from environmental samples are also continually improving and we expect that reprocessing the samples considered in this study with improved tools will result in the recovery of many additional genomes. Constructing a comprehensive genomic repository of microbial diversity is a laudable goal in itself and lays the foundation for furthering our understanding of the role of microorganisms in critical biogeochemical and industrial processes.


Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in