Selecting between two transition states by which water oxidation intermediates decay on an oxide surface
Published in Chemistry

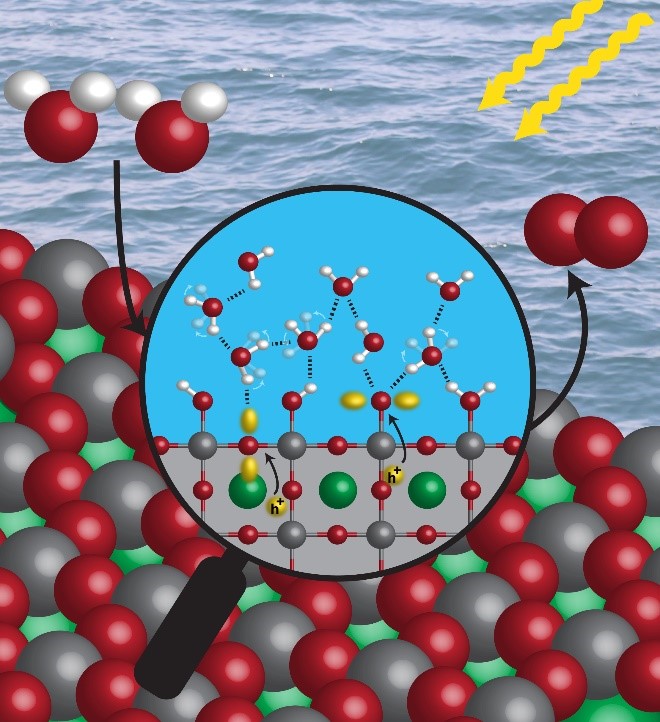
Energy storage is among the grand challenges for our society to move to a renewable energy-based economy. One way to store energy is through (photo)electrochemical catalysis, which transforms light or electrical energy into chemical bonds. This transformation occurs at electrode surfaces, and these surfaces play the role of the catalyst that reconfigures reactants into products through a series of charge transfer and chemical bond-making and breaking events. The mechanism of these transformations is intriguing and has long been the interests of our group. We asked ourselves: Can we capture these events in real-time? Can we utilize the surface properties to control the rate of catalysis such that the electrical(photo) energy can be efficiently funneled to the electrode surfaces to generate chemical products?
The answers are yes (though it took us some time to figure out). We developed in-situ time-resolved visible and infrared spectroscopies, which utilize electronic and vibrational probes to monitor catalysis. We chose heterogeneous multi-step water oxidation as our target because it plays a vital role in any (photo)electrosynthetic reactions. First, we wanted to identify the initial step for water oxidation. With our spectroscopic capability, we were able to discover Ti-O• (oxyl) and Ti-O•-Ti (bridge) species formed at ultrafast time scales (with rate 7.7×10^11/s) as initial reaction intermediates on the n-SrTiO3 electrode with near unity faradaic efficiency.
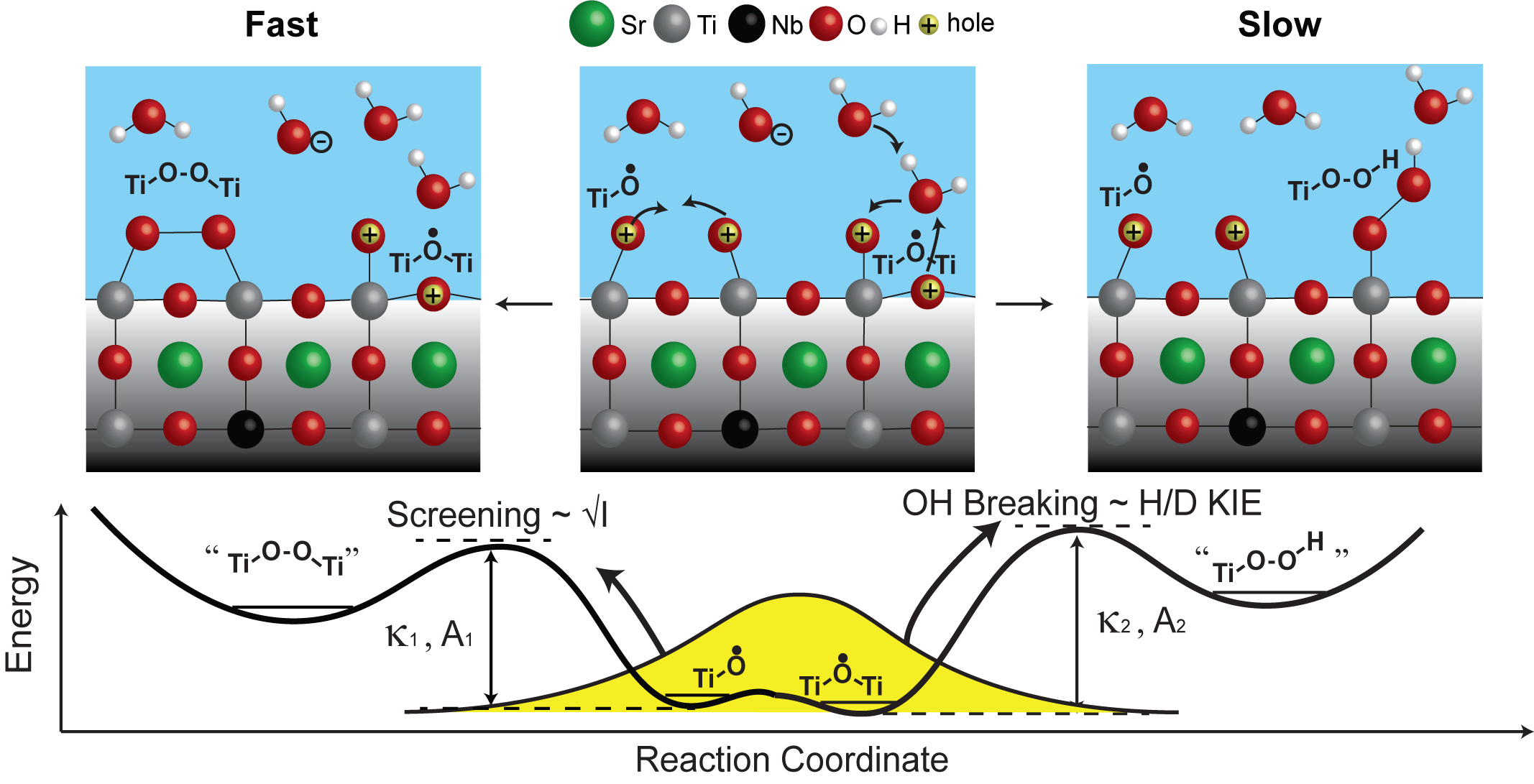
This is quite remarkable, and we took one step further to investigate how do these initial intermediates transform to the next step of the reaction. For example, the O-O bond which is a pre-requisite for O2 evolution. We discovered that there are two pathways (fast 1.25×10^5/s and slow 1.67×10^4/s) for these transformations and they depend on solution properties such as pH, ionic strength and isotopes (H vs D). To begin with, by tuning the solution pH, the population of intermediates react through the two pathways can be tuned systematically. With increasing pH, the fast pathway is favored and with decreasing pH, the slower pathway is favored. What’s more, by tuning the ionic strength and H/D isotopes, the rate of the two pathways can also be tuned. By increasing the solution ionic strength, the rate of the reaction is accelerated, and it is governed by Bronsted–Bjerrum theory. The rate of the fast pathway increased more with increasing ionic strength. Also, by changing H2O to D2O, the reaction is slowed down. The rate of the slow pathway is slowed down more with H/D isotopes.
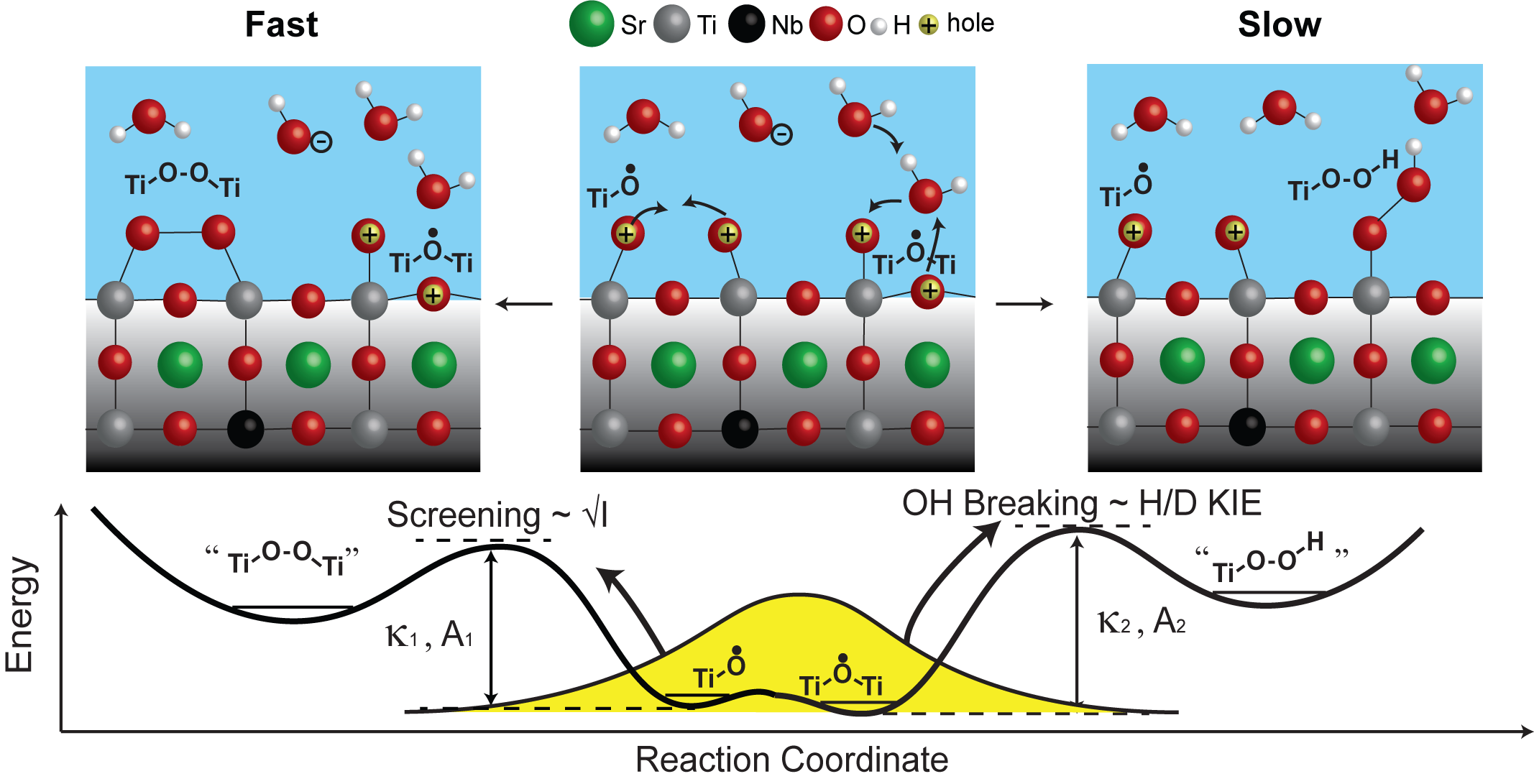
These exciting results trigger us to decipher the reaction mechanism. We propose the fast pathway to be a direct coupling of two Ti-O• (oxyl). Higher ion concentration could screen the partial positive charge on Ti-O• (oxyl) which results in faster coupling. The slow pathway to be Ti-O•-Ti (bridge) reacting with water. This process involves a breaking of OH bond, which is sensitive to H/D isotopes. Temperature dependent measurements suggest the activation barrier for both pathways to be near 0.4 to 0.5 eV. We believe with the development of our transient spectroscopy, many more complex (photo)electrochemical processes can be revealed.
For detailed information, please see our article " Selecting between two transition states by which water oxidation intermediates decay on an oxide surface" in Nature Catalysis https://www.nature.com/articles/s41929-019-0332-5
Follow the Topic
-
Nature Catalysis
This journal brings together researchers from across all chemistry and related fields, publishing work on homogeneous catalysis, heterogeneous catalysis, and biocatalysts, incorporating both fundamental and applied studies.




Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in