As a follow up to our Nature Microbiology paper, where we applied our newly developed technique - SorTn-seq - to discover regulators of CRISPR-Cas immunity, we are now pleased to publish the detailed protocol in Nature Protocols.
I developed SorTn-seq during my PhD at the University of Otago (New Zealand) under the supervision of Professor Peter Fineran. We were looking for a way to identify CRISPR-Cas regulators on a genome-wide scale, using a non-biased approach. Extensive troubleshooting and optimization went into nearly every step of the procedure, so I am thrilled to finally share all that hard work! While we used SorTn-seq to study regulation of bacterial defense systems, this technique can be applied to study the regulation of virtually any gene of interest in bacteria.
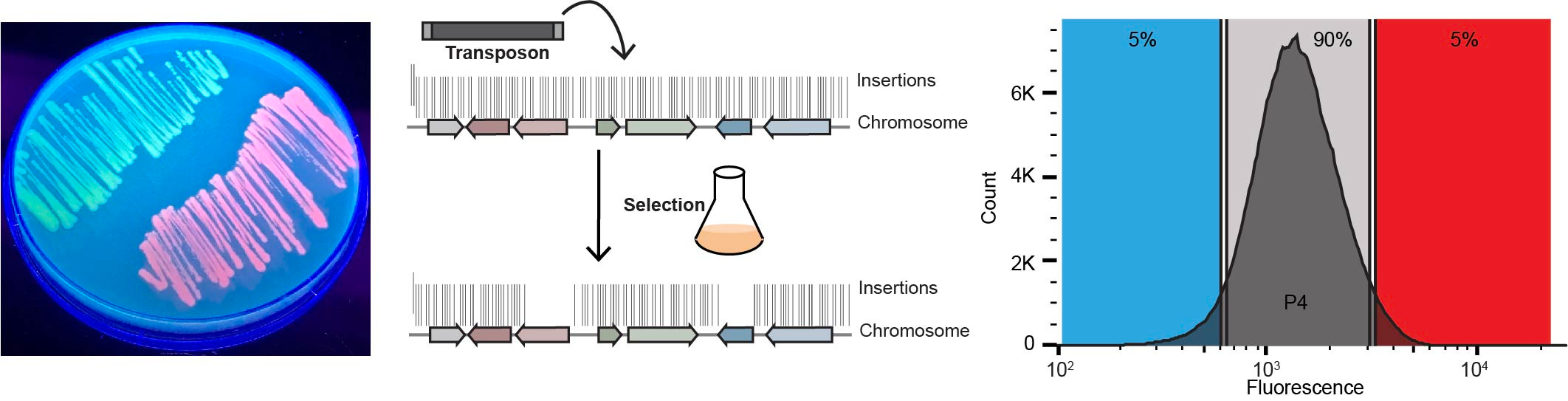
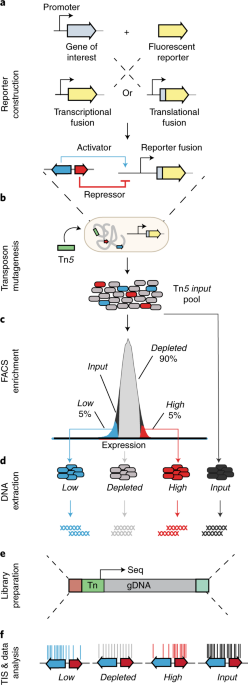
Using SorTn-seq, a high-density transposon library containing a fluorescent reporter - driven by the promoter of a gene of interest - is sorted based on fluorescence levels using cell sorting (FACS). The idea is that a random insertion within a regulator of the promoter will lead to increased or decreased fluorescence. DNA is then extracted from sorted cells to prepare sequencing libraries, which are deep-sequenced to identify the location of transposon insertions. The number of insertions within genomic features are compared amongst the different sort pools to identify factors involved in the regulation of the gene of interest (Figure 1).

Figure 1. SorTn-seq workflow. Cells harboring a fluorescent reporter are subjected to transposon mutagenesis and then sorted based on fluorescence levels. DNA is extracted from sorted cells and transposon insertion sites are identified using deep sequencing. Putative regulators are identified by comparing the transposon insertion frequency within different genomic features across sort pools (Adapted from Smith et al., 2021b).
The TraDIS pipeline (Barquist et al., 2016) is used to identify transposon tags and map reads to the reference genome. We then provide scripts for R-based data analysis (Jackson, S.A. 2021), where alignment files are used to tabulate insertion counts, which are then assigned to genomic features (based on a RefSeq general feature format (GFF) file). These features can be customized to include intergenic regions, so insertions in promoter regions or other unannotated regions can be picked up in the screen. This allowed us to identify several intergenic regions (promoters and unannotated small RNAs) implicated in CRISPR-Cas regulation (Smith et al., 2021a). Unique insertion counts per feature are then compared amongst the different sort pools - low or high expression versus the control (the depleted or “normal” expression) pool, using edgeR (Robinson et al., 2010). If a genomic feature functions as an activator or repressor of the promoter of interest, insertions within that feature should be over-represented in the high or low expression pool. Thus, the SorTn-seq approach allows us to predict regulators of target gene expression!
In addition to providing the step-by-step procedure used in our Nature Microbiology paper, this protocol also elaborates on the design of the various aspects of SorTn-seq, including fluorescent reporters, transposon mutagenesis, cell sorting, transposon insertion library preparation and sequencing, and data analysis. This information provides the reader with helpful tips to adapt the SorTn-seq technique to their own organism and gene of interest. Once fluorescent reporters and transposon mutagenesis methods have been established, the entire SorTn-seq procedure takes roughly one to two weeks.
Finally, aspects of the protocol can also be used in isolation. For example, if a researcher is only interested in transposon sequencing, our library preparation method simplifies construction through the use of commercially available kits augmented with custom PCR primers. We have successfully executed six different transposon-sequencing experiments using this library preparation method, including a recent study where we utilized gene essentiality data to predict toxin-antitoxin functionality (Hampton et al., 2020). Likewise, we used fluorescent reporters and flow cytometry in Pectobacterium carotovorum to uncover the mechanism by which an anti-CRISPR associated (Aca) protein regulates expression of its cognate anti-CRISPR (Birkholz et al., 2019).
We believe the flexibility and high-throughput nature of our functional genomics technology will now enable researchers to uncover regulators of bacterial gene expression on a genome-wide level in a single experiment. We would love for people to give the technique a try to study regulation in their favorite bacterium!
Happy Sorting!
References
Barquist, L., Mayho, M., Cummins, C., Cain, A.K., Boinett, C.J., Page, A.J., Langridge, G.C., Quail, M.A., Keane, J.A., and Parkhill, J. (2016). The TraDIS toolkit: sequencing and analysis for dense transposon mutant libraries. Bioinformatics 32, 1109-1111.
Birkholz, N., Fagerlund, R.D., Smith, L.M., Jackson, S.A., and Fineran, P.C. (2019). The autoregulator Aca2 mediates anti-CRISPR repression. Nucleic Acids Res 47, 9658-9665.
Hampton, H.G., Smith, L.M., Ferguson, S., Meaden, S., Jackson, S.A., and Fineran, P.C. (2020). Functional genomics reveals the toxin-antitoxin repertoire and AbiE activity in Serratia. Microb Genom 6.
Jackson, S.A. (2021). JacksonLab/SorTn-seq: 1.0.0 (Version v). (Zenodo).
Robinson, M.D., McCarthy, D.J., and Smyth, G.K. (2010). edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139-140.
Smith, L.M., Jackson, S.A., Malone, L.M., Ussher, J.E., Gardner, P.P., and Fineran, P.C. (2021a). The Rcs stress response inversely controls surface and CRISPR-Cas adaptive immunity to discriminate plasmids and phages. Nat Microbiol 6, 162-172.
Smith, L.M., Jackson, S.A., Gardner, P.P., and Fineran, P.C. (2021b). SorTn-seq: a high-throughput functional genomics approach to discovering regulators of bacterial gene expression. Nature Protocols.
Follow the Topic
-
Nature Protocols
This journal publishes secondary research articles and covers new techniques and technologies, as well as established methods, used in all fields of the biological, chemical and clinical sciences.

Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in