Sparse representation for machine learning the properties of defects in 2D materials
Published in Materials
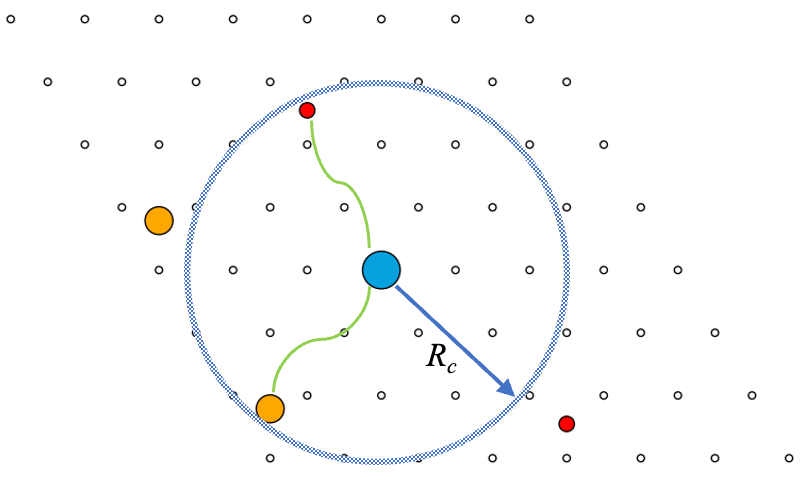

Why?
2D materials offer exciting opportunities as building blocks for new electronic devices, such as bendable screens, efficient solar panels, and high-resolution cameras. One of the defining properties of 2D materials is the high influence of crystal imperfections, or defects. They can turn isolators into semiconductors, semiconductors into metals, make materials magnetic or catalytic. They also introduce delicious flat bands. Our esteemed colleagues did a whole paper about finding materials with flat bands. I never had an idea that this is a thing, before starting to write the blog post. Thanks, Nature Communities!
Ideal crystalline materials consist of an infinite repeating pattern. Real crystalline materials have defects. In our work we study point defects: vacancies, when an atom is removed, and substitutions, when an atom is replaced with a different one. Example:
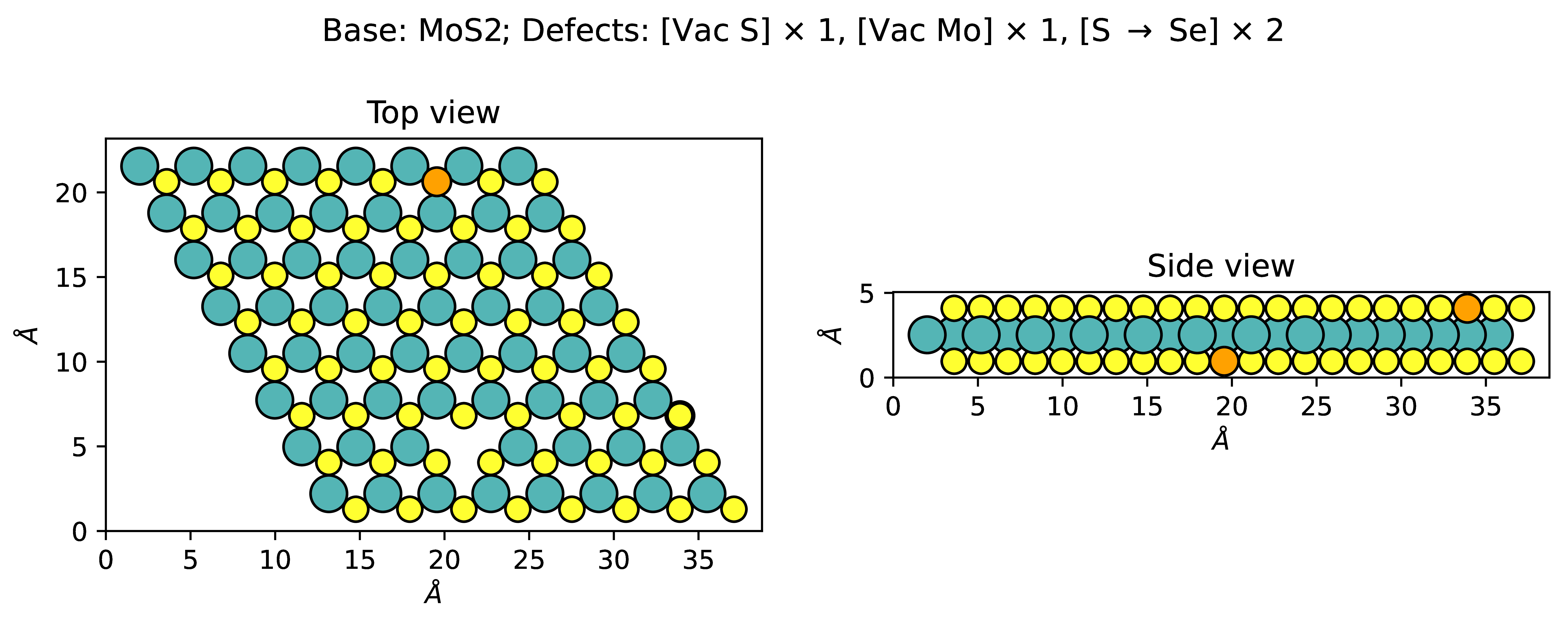
We all want to do in-silico material design. The part that makes it work is rapid estimation of the candidate material properties you must have to build your fancy generative-bayesian-differentiable-genetic models. And the cost for computational methods, such as DFT, starts at hours per structure. More unbearable than buying an apartment in Singapore as a foreigner on a university salary. Hence, machine learning: pay a bit for training structures, get 100 times more for free. The tricky part is to make machine learning actually work.
What?
In our paper we propose a really trivial and straightforward idea: when dealing with multiple point defects, input only the defects into the ML model, otherwise there is a high chance it will be worse than a pairwise potential. HowTo is in the following figure:
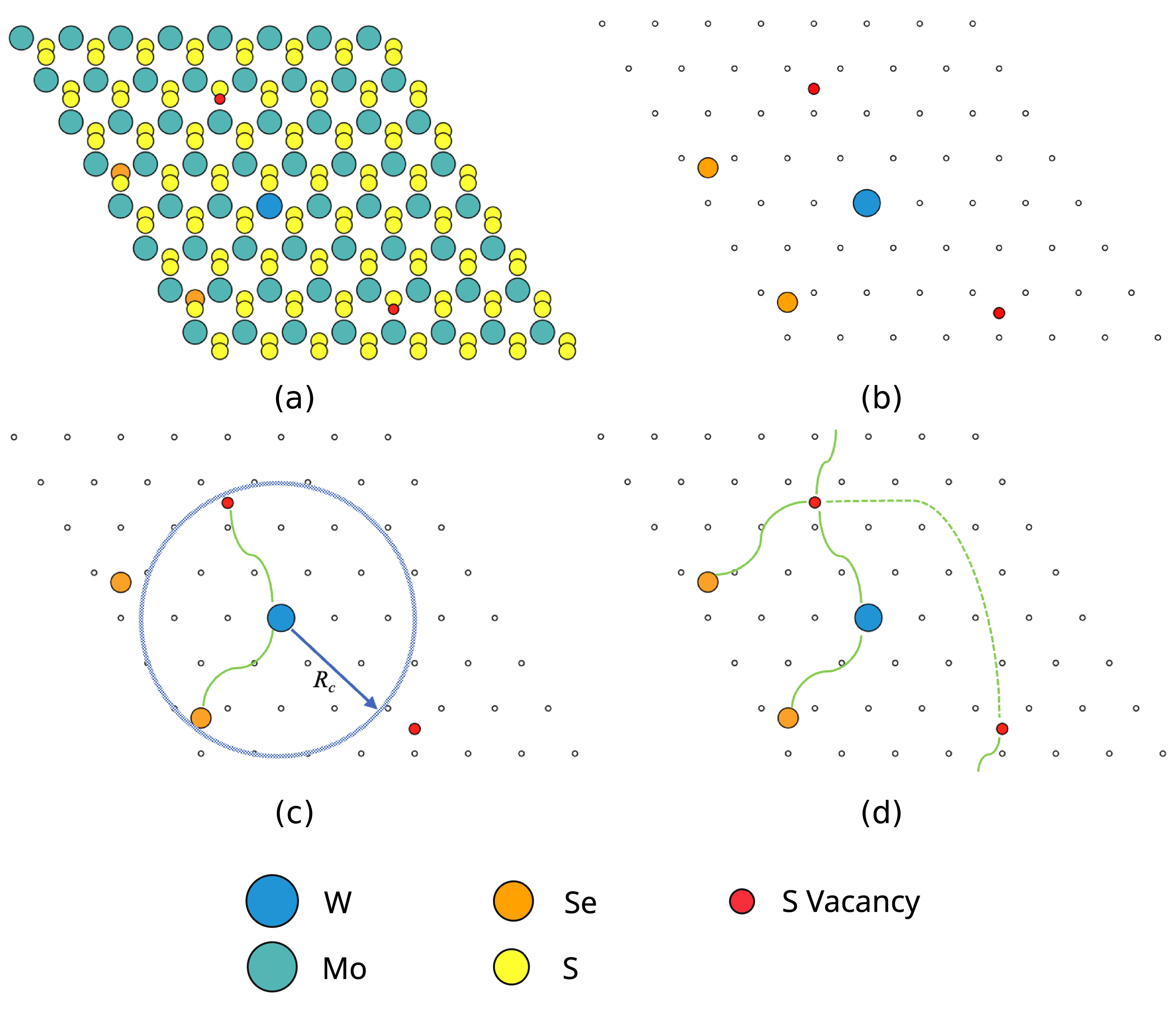
The graph with wiggly edges, and the rest of the picture, are by my dear co-author and mentor Andrey Ustyuzhanin
(a) Full structure that overwhelms the poor neural network with identical atomic neighborhoods
(b) Sparse structure, which consists only of point defects
(c) A graph built by connecting the defect sites that are closer than the cutoff radius
(d) Resulting sparse graph. Note the edges going through the periodic boundary. It’s a multigraph!
Build a graph of defects, add the base material chemical formula as a global property, and feed to your favorite graph neural network. Profit! 3.7 times less mean absolute error than the next best method.
Can I have my quantum computer now?
No, you may not. Theoretically, with right defect engineering we can tailor-make materials for everything, including qubits. Practically, there are three big limitations:
- We don’t predict defect migration and stability. Experimentally, vacancies tend to congregate into big distinctly non-quantum holes. The primary concern here is the training data, computing finite-temperature ab initio MD is very expensive.
- Really fancy properties (will this work as a qbit?) are complicated to compute (e. g. quantum MC) both in terms of computing power and expert tuning for each structure. Training data are again a problem.
- Generalizing to new materials. It’s not a principled limitation, and could be addressed by using any of the fancy GNNs out there for material representation together with our sparse representation for defects. We may write a paper about it. Or not. Academic career is such a precarious thing. So many people just go crazy.
The juicy drama bits. It’s “a behind the paper” post after all
Paper happened by accident. We wanted to skip straight to material design, but found out that general-purpose structure-property graph neural networks basically don’t work for structures with multiple point defects. Idea for your next paper: do some clever ML trick to make them, prove our paper redundant.
I had a very pleasant interaction with one of the reviewers. On the first submission, they wrote a brief paragraph about how our work is limited and useless. I fully agreed. On the second, they recommend acceptance without further modification. I don’t know who you are, but I love you! Take that, PhD comics!
Computational research projects evolve into software engineering projects. The core idea of the paper was invented and tested in about a week. All the remaining 1.5 years went into writing the code to generate the data, writing the code to process it, writing the code to implement baselines, writing the code to do hyperparameter search (which didn’t change the conclusions in the slightest, we still won by a large margin). And then running, debugging and re-running. Would be nice if there was a boilerplate library. The code had to be runnable at 4 different HPC systems, only one of which supported using own Docker containers. And python package management is horrible when CUDA versions get involved.
Data availability
https://research.constructor.tech/open/2d-materials-point-defects
Code availability
Code: https://github.com/HSE-LAMBDA/ai4material_design; it can be run online at at https://research.constructor.tech/p/2d-defects-prediction (robs you of the joy of compiling pytorch-geometric, but you’ll have fun in other ways).
Follow the Topic
-
npj Computational Materials
This journal publishes high-quality research papers that apply computational approaches for the design of new materials, and for enhancing our understanding of existing ones.

Related Collections
With Collections, you can get published faster and increase your visibility.
Altermagnetic Materials: Theory, Simulation, and Renewed Perspectives
Publishing Model: Open Access
Deadline: Feb 28, 2027
Recent Advances in Active Matter
Publishing Model: Open Access
Deadline: Sep 01, 2026


Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in