Stereoselective conjugate cyanation of enals by combining photoredox and organocatalysis
Published in Chemistry

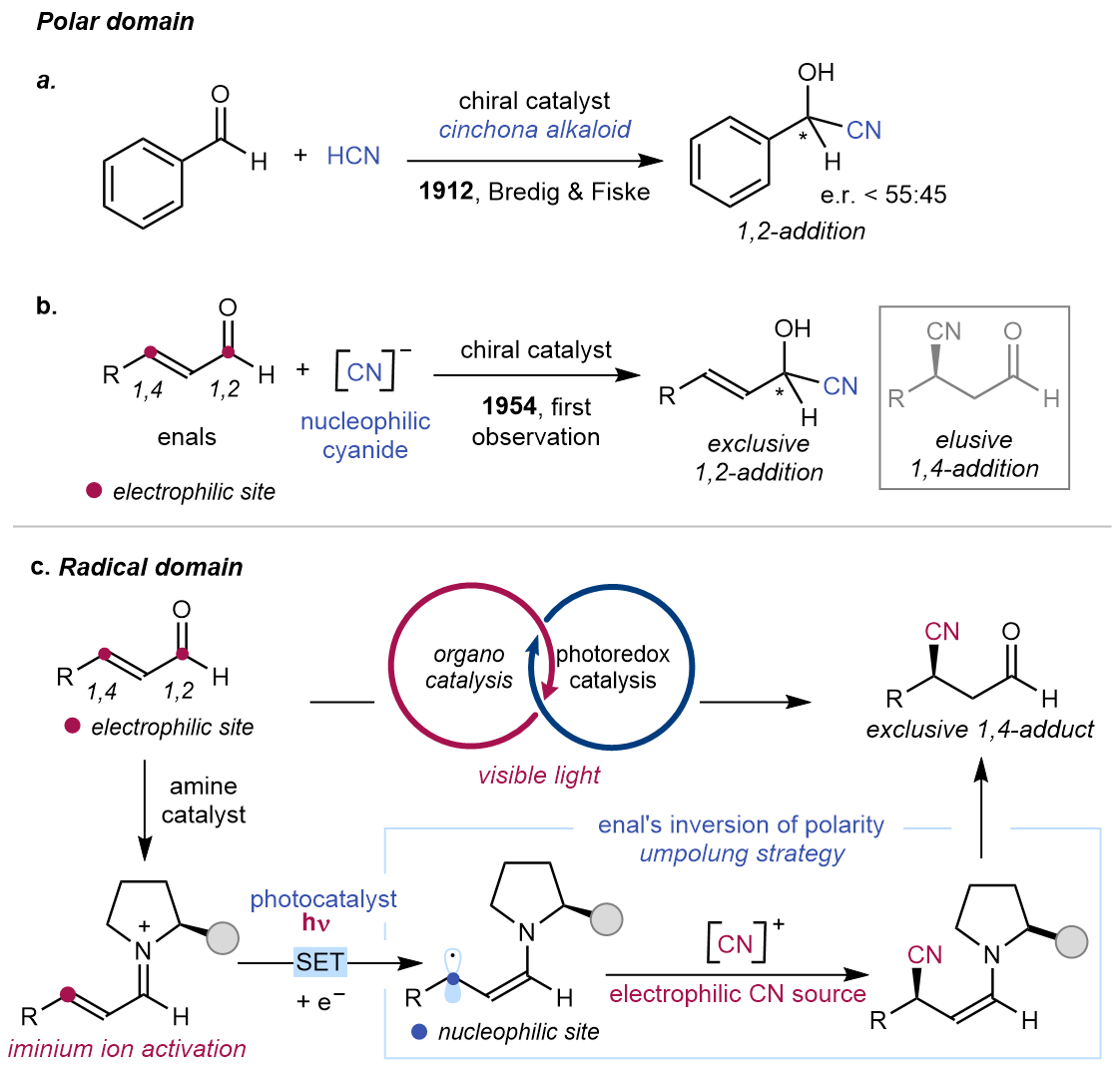
Being one of the first asymmetric organocatalytic reaction ever reported1, the asymmetric cyanation of carbonyl compounds has been extensively studied2,3. Although examples for 1,2-cyanation of α,β-unsaturated aldehydes exist, the parent 1,4-cyanation is complicated by chemoselectivity issues: as Prelog and Wilhelm described, combination of nucleophilic cyanide and enals delivered the 1,2-addition cyanohydrin as the sole product4. Because of electronic and steric factors, nucleophilic cyanide has an overwhelming preference for the carbonyl carbon of enals. To date, there is no general 1,4-cyanation procedure that can override the intrinsic selectivity for enals through polar pathways5.
We were intrigued by the longstanding challenge in organic synthesis, and approached the problem with a “radically” different strategy. A radical cyanation was conceptualized, using an electrophilic cyanide source and a 5π-enaminyl radical, which was previously reported to be nucleophilic in nature6. Informed by our earlier investigations on photochemistry of chiral iminium ions7, we reasoned that their electron-poor nature could be leveraged for a facile single electron transfer (SET) reduction to form a chiral nucleophilic radical, which then can be intercepted in asymmetric fashion with electrophilic tosyl cyanide, an established regent for radical nitrile transfer8. By reversing the polarity of the enal component, the cyano group transfer to the β-position of an enal should unlock a solution to this problem and enable further transformations by such cross electrophile coupling approach.
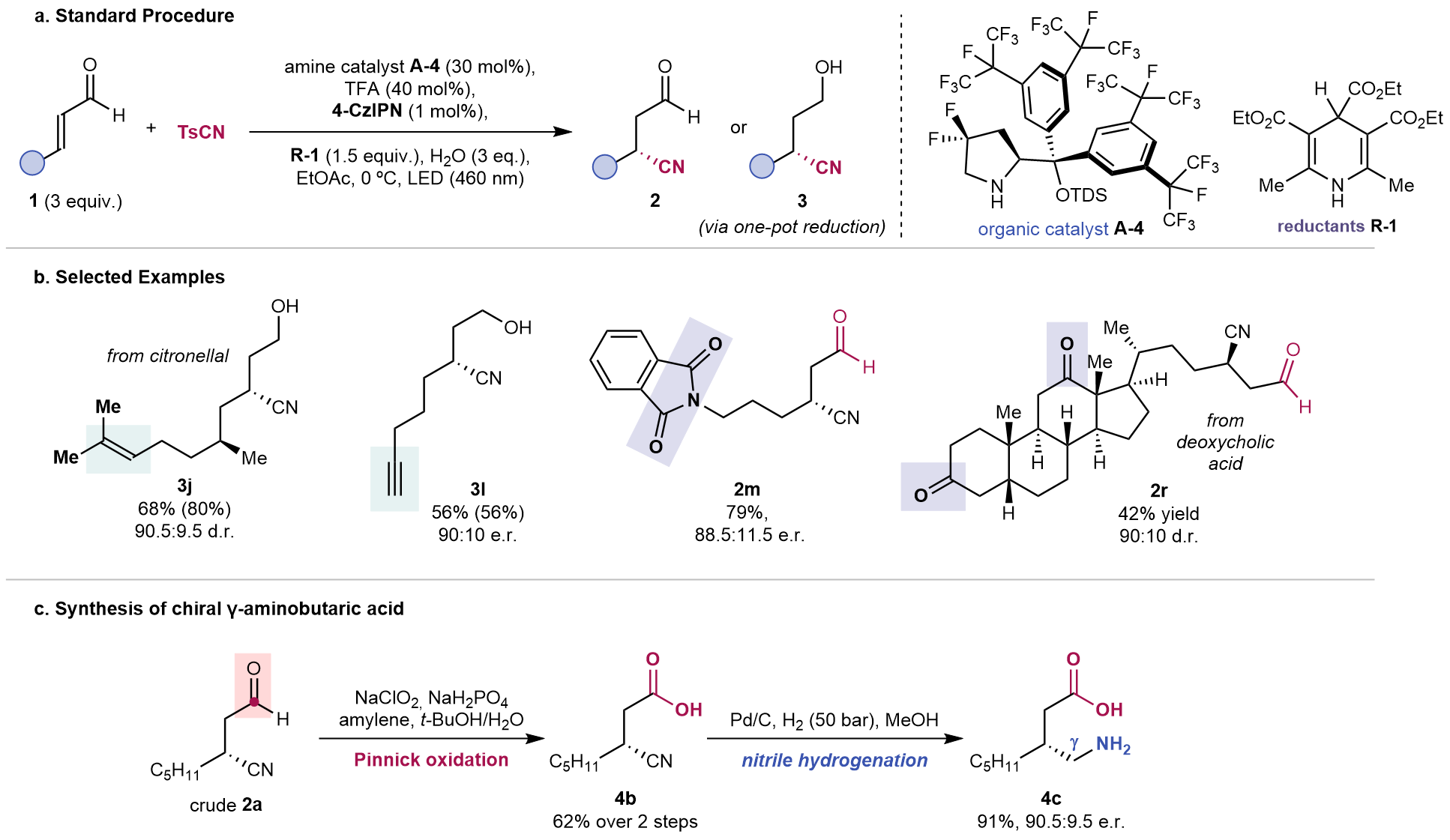
We set out to explore the idea using our difluorinated pyrrolidine catalyst, which is amenable to radical asymmetric iminium ion catalysis7, an external photocatalyst and a 1,4-dihydropyridine as sacrificial reductant. We were pleased to see the β-cyanoaldehyde product formed with exclusive 1,4-chemoselectivity and good enantioselectivity over a variety of aliphatic enals bearing diverse functional groups, such as alkyne and phthalimide. More complex enals, including natural product derived scaffolds, could also be transformed, without altering the pre-installed alkene and ketone functionalities. Furthermore, stereoinduction of the cyanation is dictated by the aminocatalyst and not influenced by the pre-existing stereocenters in the starting materials. The cyanoaldehyde products are of great synthetic value, as shown in the short preparation of an unnatural chiral γ-amino acid after redox manipulation steps.
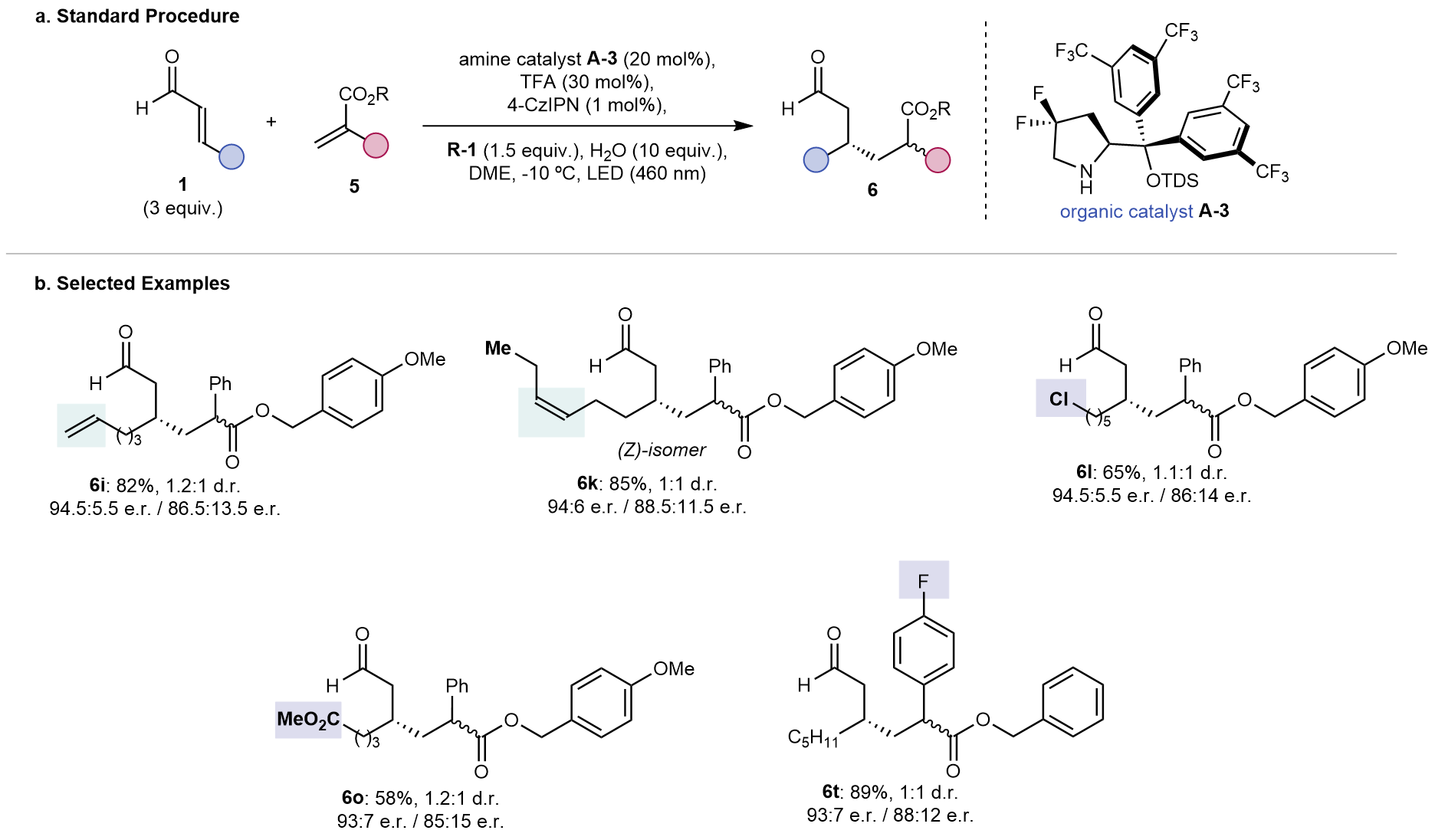
We then searched for other reactivities using this umpolung platform, such as a cross-electrophile coupling of two Michael acceptors to form synthetically challenging linear 1,6-dicarbonyl compounds9. Indeed, the reaction delivered the desired product with consistently good yields and high level of enantioinduction. A similar level of functional group tolerance as observed for the β-cyanation was also found, since reactive handles such as alkyl chloride, ester and aryl fluoride were unaffected. Terminal and internal alkenes, which often participate in radical transformations, were retained without double bond isomerization. Although the products were obtained as mixture of inseparable diastereomers, they could be separated upon global reduction to the corresponding alcohols.
In conclusion, we demonstrated that the synergy of photoredox catalysis and asymmetric organocatalysis can be a general, versatile strategy for exerting enantiocontrol over novel radical reactions. Encouraged by the initial success, we believe that there is unexplored potential with our current methods. Investigations to extend this concept is currently in progress.
References
- Bredig, G. & Fiske, P. S. Beiträge zur chemischen Physiologie und Pathologie. Biochem. Z. 46, 7-23 (1912).
- Kurono, N. & Ohkuma, T. Catalytic asymmetric cyanation reactions. ACS Catal. 6, 989–1023 (2016).
- Zhou, H., Zhou, Y., Bae, H. Y., Leutzsch, M., Li, Y., De, C. K., Cheng, G.-J., List, B. Organocatalytic Stereoselective Cyanosilylation of Small Ketones. Nature, 605, 84–89 (2022).
- Prelog, V. & Wilhelm, M. Untersuchungen über asymmetrische Synthesen VI). Der Reaktionsmechanismus und der sterische Verlauf der asymmetrischen Cyanhydrin-synthese. Helv. Chim. Acta. 37, 1634–1660 (1954).
- Zeng, X.-P., Sun, J.-C., Liu, C.-, Ji, C.-B. & Peng, Y.-Y. Catalytic asymmetric cyanation reactions of aldehydes and ketones in total synthesis. Adv. Synth. Catal. 361, 3281–3305 (2019).
- Terrett, J. A., Clift, M. D. & MacMillan, D. W. C. Direct β-alkylation of aldehydes via photoredox organocatalysis. J. Am. Chem. Soc. 136, 6858–6861 (2014).
- Silvi, M., Verrier, C., Rey, Y. P., Buzzetti, L. & Melchiorre, P. Visible-light excitation of iminium ions enables the enantioselective catalytic β-alkylation of enals. Nat. Chem. 9, 868–873 (2017).
- Barton, D.H.R., Jaszberenyi, J. & Theodorakis, E.A. The invention of radical reactions. part XXIII new reactions: Nitrile and thiocyanate transfer to carbon radicals from sulfonyl cyanides and sulfonyl isothiocyanates. Tetrahedron, 48, 2613 (1992).
- Seebach, D. Methods of reactivity umpolung. Angew. Chem. Int. Ed. 18, 239–258 (1979).
Follow the Topic
-
Nature Catalysis
This journal brings together researchers from across all chemistry and related fields, publishing work on homogeneous catalysis, heterogeneous catalysis, and biocatalysts, incorporating both fundamental and applied studies.





Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in