Supported catalysts deactivate by decomposing into atomic species
Published in Chemistry
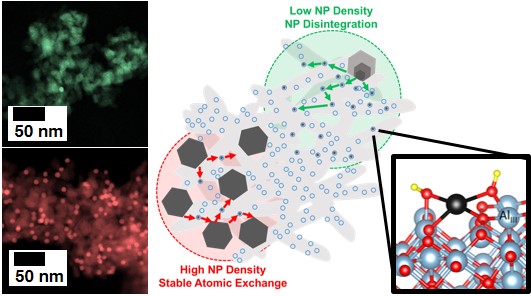

Nearly all supported metal catalysts lose activity over time. The most common cause of catalyst deactivation is loss of reactive surface area; typically, this is chalked up to the growth of large crystallites at the expense of smaller crystallites, leading to an overall decrease in exposed metal sites. In this work, we reveal the opposite process as a stealthier – but equally severe - deactivation mechanism. Instead of the emergence of large metal aggregates, particles disappear into ensembles of single atoms and nanoclusters. Unfortunately, the extent of this novel deactivation mechanism is difficult to quantify in most catalytic systems, due to the difficulty in identifying such highly disperse species among many others. Nevertheless, we demonstrate that nanoparticle redispersion is a phenomenon which may affect catalyst activity at rates 100 times faster than particle growth. How can we predict if particle growth, or particle redispersion will dominate the stability of a catalyst? The size and spatial distributions of nanoparticles hold the key!
However, in typical catalyst synthesis, it is difficult, if not impossible, to control these two parameters. In this contribution, we utilize colloidal synthesis techniques to independently control nanoparticle size and spatial distribution on model Pd/Al2O3 emissions-control catalysts. Techniques to control the synthesis of Pd nanoparticles, within a single nanometer precision, have been developed in our lab over the last few years. Then, to change the spatial distribution of nanoparticles, we simply varied the quantity of preformed nanoparticles added to the Al2O3 support. The stability results were surprising and counterintuitive: where high nanoparticle loadings proved completely stable in aging experiments, sparse nanoparticle loadings exhibited rapid and severe deactivation. This loading-dependent stability held across different nanoparticle sizes, as well as polydisperse materials, although the specific temperature of particle redispersion varied with particle size. Such an inverse dependence of catalyst stability on spatial distribution, where closer nanoparticles are more stable, contradicts the general understanding in the field, where more isolated particles are more stable. This mechanism is particularly surprising on the commercially relevant, unmodified Al2O3 support. Although the formation of single atomic species is often described on reducible oxide supports such as CeO2, few reports describe such atomic species on Al2O3. Here, density-functional theory demonstrates why this is the case - the formation of any atomic Pd species on Al2O3 require significant energies of formation. However, at high enough temperatures, this is possible! In agreement with aging experiments, calculations suggest that the Pd(OH)2 atomic adsorbate, tethered to AlIII defect sites, are the most likely formed atomic species. Using a thermodynamic model and quantifying the ratio between Pd atoms and AlIII defect sites, we explain how atomic Pd species may be formed on commercial Al2O3 supports, and how this mechanism is more prevalent at lower nanoparticle densities. Overall, this work shows how colloidal catalyst synthesis can be used to deconvolute the effects of size and loading on catalyst stability, and demonstrates that for some reactions, higher nanoparticle loadings may be more stable than spatially isolated nanoparticles.
Follow the Topic
-
Nature Catalysis
This journal brings together researchers from across all chemistry and related fields, publishing work on homogeneous catalysis, heterogeneous catalysis, and biocatalysts, incorporating both fundamental and applied studies.


Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in