Switching from immunosuppression to tolerance via controlled delivery
Published in Bioengineering & Biotechnology

Type I diabetes (T1D) is a metabolic disorder characterized by an autoimmune attack on insulin-producing beta islet cells, resulting in impaired glycemic control. Islet transplantation is a potentially beneficial therapy for the T1D population. During this procedure, exogenous islets are infused via the portal vein into the liver. Given that these islets are usually allogeneic, immunosuppressive therapy is required. A plethora of side effects are associated with immunosuppression, and therefore islet transplantation is only utilized for patients that have experienced severe cases of hypoglycemia or for patients already on immunosuppressive therapy for another transplant.
Immunosuppressive drugs tend to act on pathways with cell-type-specific effects. Traditionally, the target for immunosuppressive drugs, like rapamycin, is T cells. These therapies aim to arrest T cell proliferation but knocking out T cells leaves patients vulnerable to infection and cancer. Due to the challenges of selective drug delivery to only T cells, immunosuppressants reach a variety of cell types and generate a wide range of side effects. Our objective was thus to allow for islet graft acceptance without depleting a patient’s immunological defenses and with minimal off-target effects.
Rapamycin is a maintenance immunosuppressive drug, prescribed for a variety of transplantation procedures. Rapamycin functions by inhibiting the mTOR pathway. While mTOR regulates T cell proliferation, it has many other functions depending on the cell type. For example, inhibiting mTOR in hepatocytes causes an upregulation of gluconeogenesis. This causes glucose to be readily released from these cells, resulting in metabolic and lipid dysregulation. We aimed to deliver rapamycin in a way that would prevent delivery to off-target cells and thus reduce such side effects (Fig. 1).

Figure 1 | Inhibition of mTOR by rapamycin has cell-type-specific effects. In T cells, mTOR inhibition by rapamycin arrests T cell proliferation and inflammatory cytokine secretion. In antigen-presenting cells, mTOR inhibition by rapamycin induces a tolerogenic phenotype, characterized by a lack of expression of costimulatory receptors. In hepatocytes, mTOR inhibition by rapamycin enhances gluconeogenesis to cause metabolic and lipid dysregulation.
Of note, we understood that rapamycin has tolerogenic effects on antigen-presenting cells. Our research team has developed a drug delivery platform consisting of polymeric nanoparticles that can be customized for targeting specific cell types. The nanoparticles are formed from poly(ethylene glycol)-b-poly(propylene sulfide) (PEG-b-PPS) copolymers. The nanoparticles self-assemble due to interactions between aqueous solvents and the hydrophilic PEG block and the hydrophobic PPS block. Based on the PEG fraction of the polymer, a variety of different nanostructures can be formed (Fig. 2).
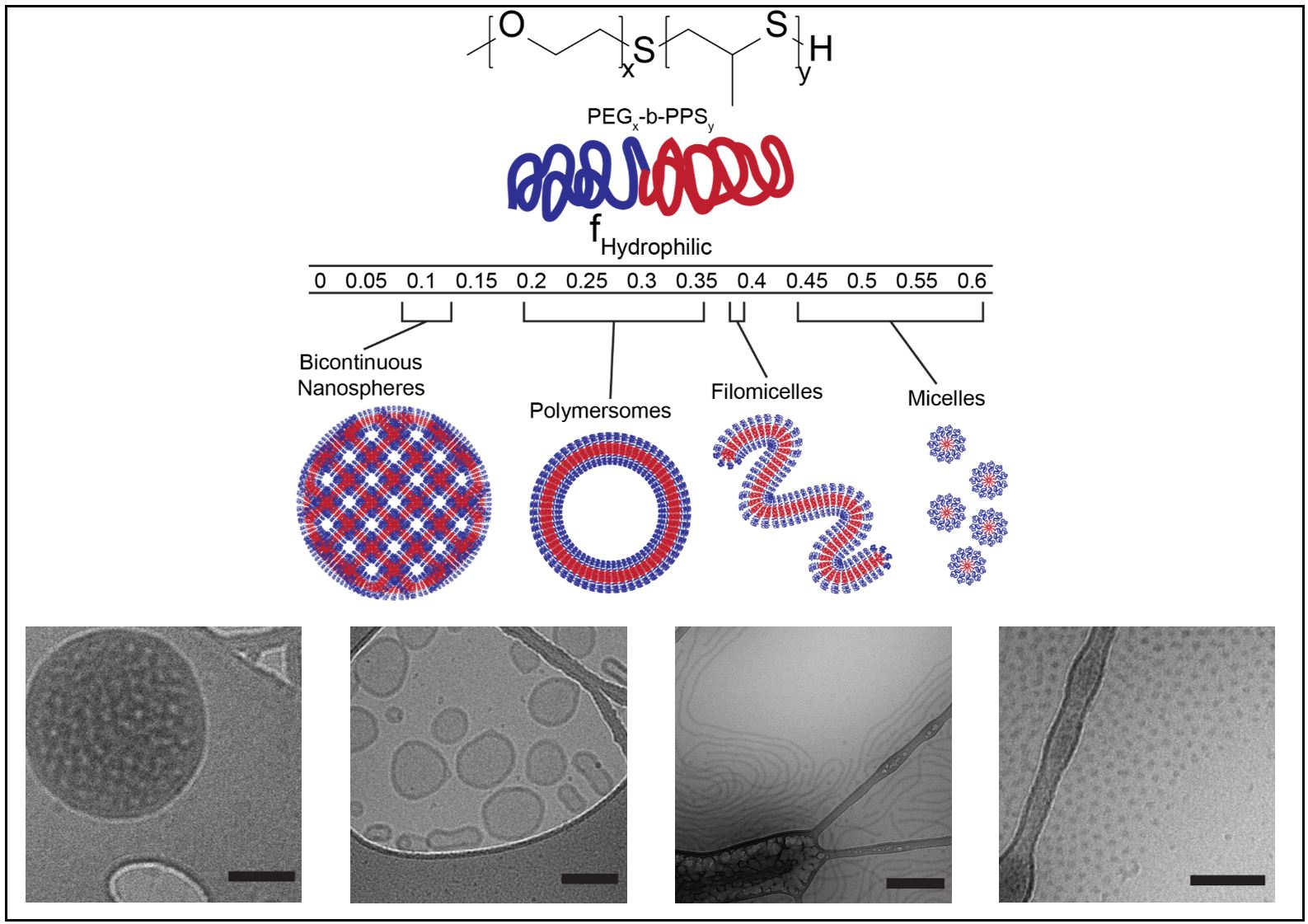
Figure 2 | Poly(ethylene glycol)-b-poly(propylene sulfide) copolymers self-assemble into a variety of nanostructures, each with unique properties. Top: Chemical structure of poly(ethylene glycol)-b-poly(propylene sulfide) (PEG-b-PPS) copolymers. Middle: Schematic showing how the assembled nanostructure can be specified by varying the length of the hydrophilic PEG fraction of the copolymer. Bottom: Cryogenic transmission electron (cryoTEM) images of nanostructures. From left to right: bicontinuous nanospheres, polymersomes, filomicelles, micelles.
Each nanostructure is selectively uptaken by a unique cell type in a process we refer to as Nanostructure-Enhanced Targeting (NSET). The vesicular polymersome nanostructures show a preference for uptake by antigen-presenting cells, including macrophages and dendritic cells.
While difficult to deliver via the parenteral route of administration due to poor water solubility, rapamycin can be easily loaded into the hydrophobic PPS bilayer of polymersomes. Entrapment within our nanocarriers prevents rapamycin metabolism and enables targeting of antigen-presenting cells. Phagocytosis of our rapamycin-loaded polymersomes (rPS) by these cells allows intracellular release of the drug and induction of a tolerogenic phenotype. Subcutaneous injection of rPS permits lymphatic drainage and facile access to high concentrations of antigen presenting cells in lymph nodes (Fig. 3).
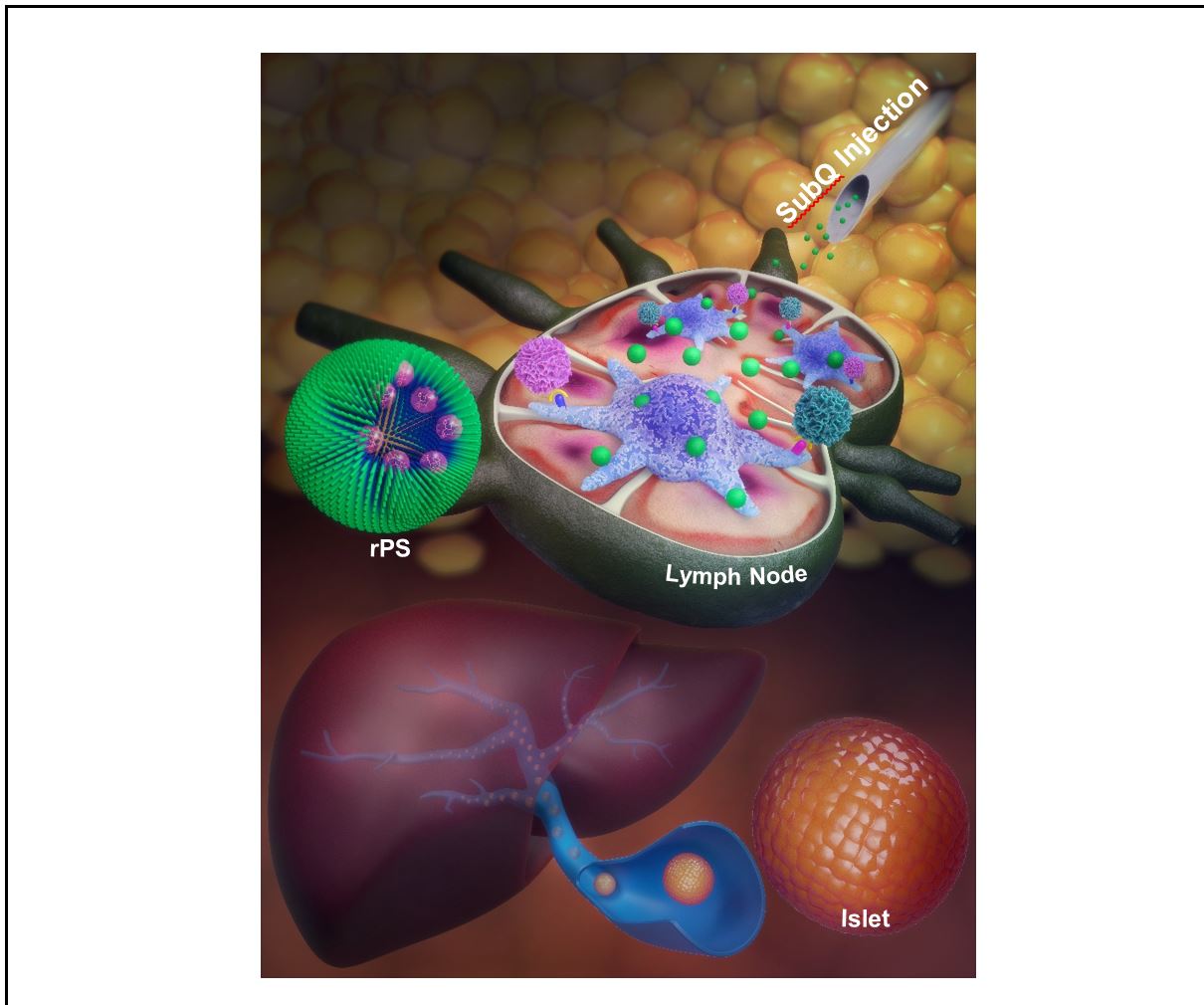
Figure 3 | Subcutaneous injection of rapamycin-loaded polymersomes allows for lymphatic drainage and targeting of antigen presenting cells, which can tolerize T cells to allogeneic islets. Illustration by Alex J. Jerez.
Tolerogenic antigen-presenting cells “talk” to T cells via cell-cell interactions to selectively inhibit their activity. Specifically, these antigen-presenting cells fail to express costimulatory molecules, including CD40, CD80, and CD86, resulting in a potent costimulation blockade. This causes T cells to go into an inactive state known as anergy.
The holy grail of transplantation immunotherapy is a drug that can induce antigen-specific tolerance. By using rPS to concentrate rapamycin delivery to antigen-presenting cells (Fig. 4), we avoid the non-specific systemic immunosuppression and side-effects that respectively result from rapamycin uptake by T cells and off-target cell types. By timing the delivery of the tolerance-inducing rPS with the injection of allogeneic islets, we show that cellular targeting via engineered nanostructures can be used to achieve antigen-specific tolerance. A mixed lymphocyte reaction (MLR) assay verified the suppression only of T cells specific to antigens found in islets from donor mice, while T cell activation was induced following exposure to non-donor antigens.
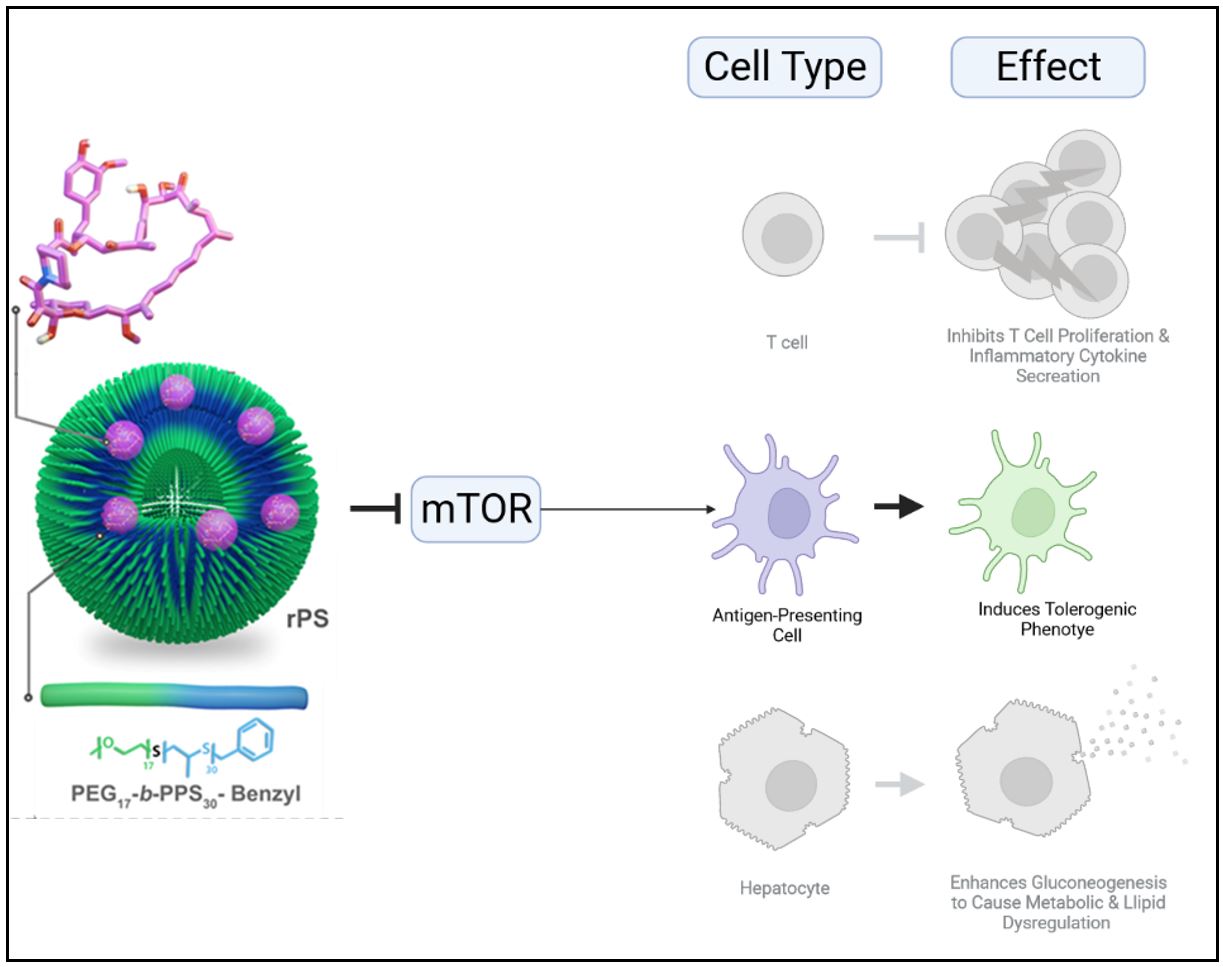
Figure 4 | Targeted delivery of rapamycin to antigen-presenting cells confers tolerance and reduces side effects. In T cells, mTOR inhibition by rapamycin arrests T cell proliferation and inflammatory cytokine secretion. In antigen-presenting cells, mTOR inhibition by rapamycin induces a tolerogenic phenotype, characterized by lack of expression of costimulatory receptors. In hepatocytes, mTOR inhibition by rapamycin enhances gluconeogenesis to cause metabolic and lipid dysregulation. By using rapamycin-loaded polymersomes (rPS), targeted delivery of rapamycin can be used to induce antigen-specific inhibition of T cells to achieve therapeutic immunosuppression without impacting other cell types.
In addition to achieving antigen-specific tolerance, we found that rPS treatment reduces side effects common to rapamycin treatment, including alopecia. RNA sequencing of T cells demonstrated that rPS treatment avoids transcriptional level changes associated with infection, malignancy, and metabolic syndrome.
Going forward, we have many unanswered questions. We have carefully studied the allogeneic response in a fully-major histocompatibility complex mismatched and clinically relevant intraportal islet transplantation mouse model. However, type I diabetics also have autoimmune responses that must be suppressed. This leads us to the question “do rPS tolerize against autoimmune responses?” We hope to investigate this question in a nonobese diabetic mouse model.
Furthermore, given the niche nature of islet transplantation, especially in the United States, we seek to investigate if rPS therapy will provide tolerogenic benefits for other types of transplantations. We hope to investigate kidney transplantation due to the widespread implications of this therapy and the lack of an autoimmune component.
Follow the Topic
-
Nature Nanotechnology
An interdisciplinary journal that publishes papers of the highest quality and significance in all areas of nanoscience and nanotechnology.


Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in