Tandem catalysis enables energy- and carbon-efficient CO2 electroreduction to multicarbon products
Published in Chemistry
The electrochemical CO2 reduction reaction (CO2RR) provides an approach to low-carbon-intensity electrified production of chemicals and fuels using renewable electricity. CO2RR electrolyzers have, until now, relied on neutral and alkaline conditions to achieve high activity and selectivity. In these conditions, though, over 75% of the input CO2 is lost chemically as it reacts with hydroxyl ions (OH⁻) to form (bi)carbonates due to high local alkalinity in the cathode. The result is low utilization and conversion of the input CO2 feedstock: these do not achieve high single-pass CO2 conversion efficiency (SPCE), and this adds an energetic penalty to recover the lost CO2 reactants.
We report a tandem catalysis strategy that suppresses the hydrogen evolution reaction (HER) and promotes the efficient conversion of CO2 into multicarbon products (e.g., ethylene, ethanol, n-propanol) in an acidic electrolyte. This allows us to make progress against the problem of low CO2 utilization in electrocatalysis.
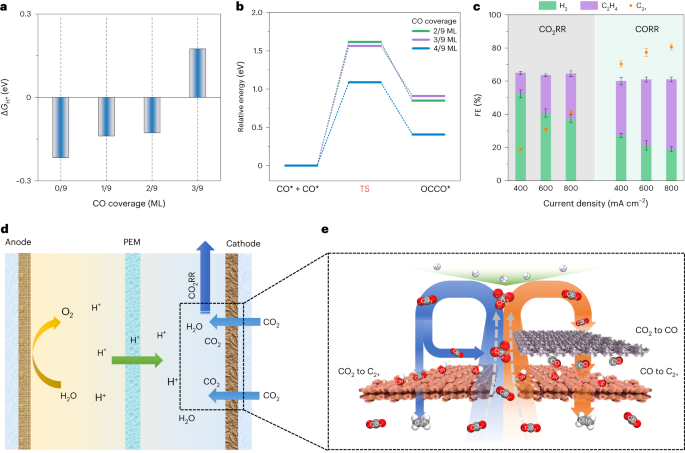
Our investigation began with the observation that a high CO coverage on the catalyst surface reduces the Gibbs free energy of H adsorption, thus suppressing HER. Additionally, we found that increased CO coverage lowers the energy barrier for C-C coupling, a critical step for the formation of multicarbon products. These results motivated us to seek routes to generate a high local concentration of CO on the catalyst surface.
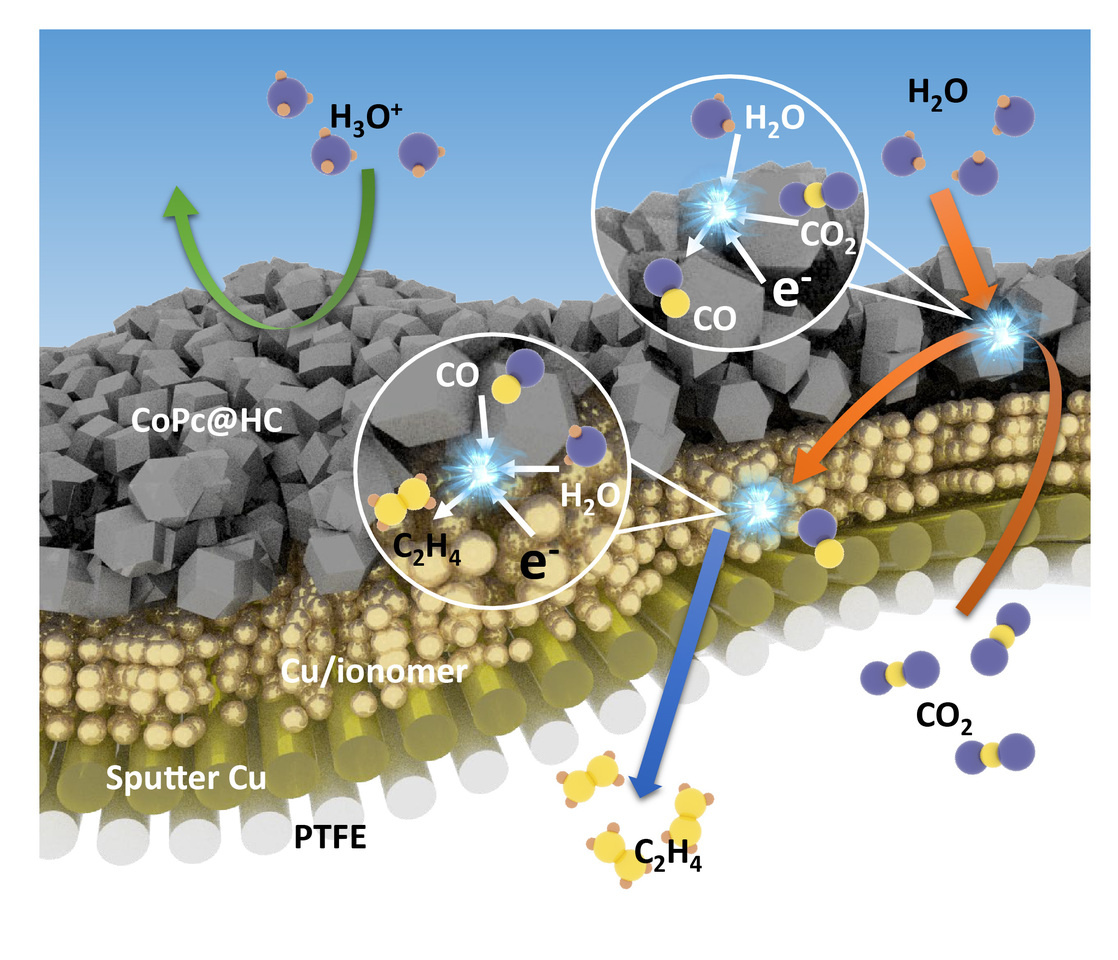
We sought therefore to decouple the CO2-to-C2+ reaction into two steps: CO2-to-CO; followed by CO-to-C2+. This, we reasoned, would allow us to increase local CO availability and create a CO-rich reaction environment (Figure 1a and 1b). Noting that the two distinct steps rely on very distinct catalyst properties, we pursued a tandem catalysis strategy, employing one catalyst optimized for the first chemical transformation CO2-to-CO, and a distinct, yet closely positioned, second catalyst, for CO-to-C2+.
Our first challenge was to design a highly selective and stable CO2-to-CO catalyst in acidic media. Catalysts that were known to be effective in alkaline and neutral electrolytes exhibited poor selectivity and rapidly declined in performance when transferred to acidic conditions, this the result of catalyst agglomeration and restructuring. To overcome this, we developed a catalyst based on atomically dispersed cobalt phthalocyanine (CoPc) on a strongly-interacting hollow carbon support (CoPc@HC). This showed CO Faradaic efficiency (FE) of 94% and H2 FE of < 3% at 300 mA cm-2, and it maintained performance over extended operating periods.
We then turned our attention to the tandem catalyst, one that would unite the CO2-to-CO and the C-C coupling steps on a single electrode. We observed that CO, with its low solubility in aqueous electrolyte, would not dissolve efficiently therein, thus leading to a CO concentration gradient from the CO-producing layer to the C-C coupling catalyst layer. This gradient could drive CO downward through the C-C coupling catalyst layer, favoring the desired reaction sequence and achieving local enrichment.
The tandem electrode therefore consisted of a three-dimensional Cu/ionomer interface catalyst layer for the C-C coupling step, followed by an upper CoPc@HC catalyst layer for the CO2-to-CO step (Figure 1c). The CoPc@HC/Cu tandem electrode achieved a C2H4 FE of 54% and a C2+ FE of 80% at 800 mA cm-2. In contrast, the Cu electrode without the tandem configuration displayed FE for C2H4 of 27% and for C2+ of 41%. Additionally, the tandem electrode exhibited <10% H2 FE compared with >35% in the case of the Cu electrode.
We observed that the sub-nanometer-thick spaces between CoPc@HC and Cu catalyst layers reduce the energy required for C-C coupling and regulate the reaction pathway to favor C2H4 production. With the aim of applying this finding to enhance C2H4 production, we further increased the spacing between CoPc@HC and Cu particles by constructing a CoPc@HC/(Cu+CoPc@HC) tandem electrode. This led to a 61% C2H4 FE and an 82% C2+ FE at 800 mA cm-2 while maintaining <10% H2 FE. When optimized for single-pass utilization, the system reaches an SPCE of 90% ± 3%, simultaneous with 55% ± 3% C2H4 FE and a total C2+ FE of 76% ± 2%.
The strategy enables energy- and carbon-efficient CO2 electroreduction to produce multicarbon products via the union of high CO2 utilization, high electron utilization (FE), and high activity (partial current density)(Figure 1d). Technoeconomic analysis suggested this tandem electrode system can offer a lower energy cost for C2H4 and C2+ production in comparison to relevant prior alkaline/neutral/acidic systems.
The full paper can be found here:
https://www.nature.com/articles/s41565-023-01543-8
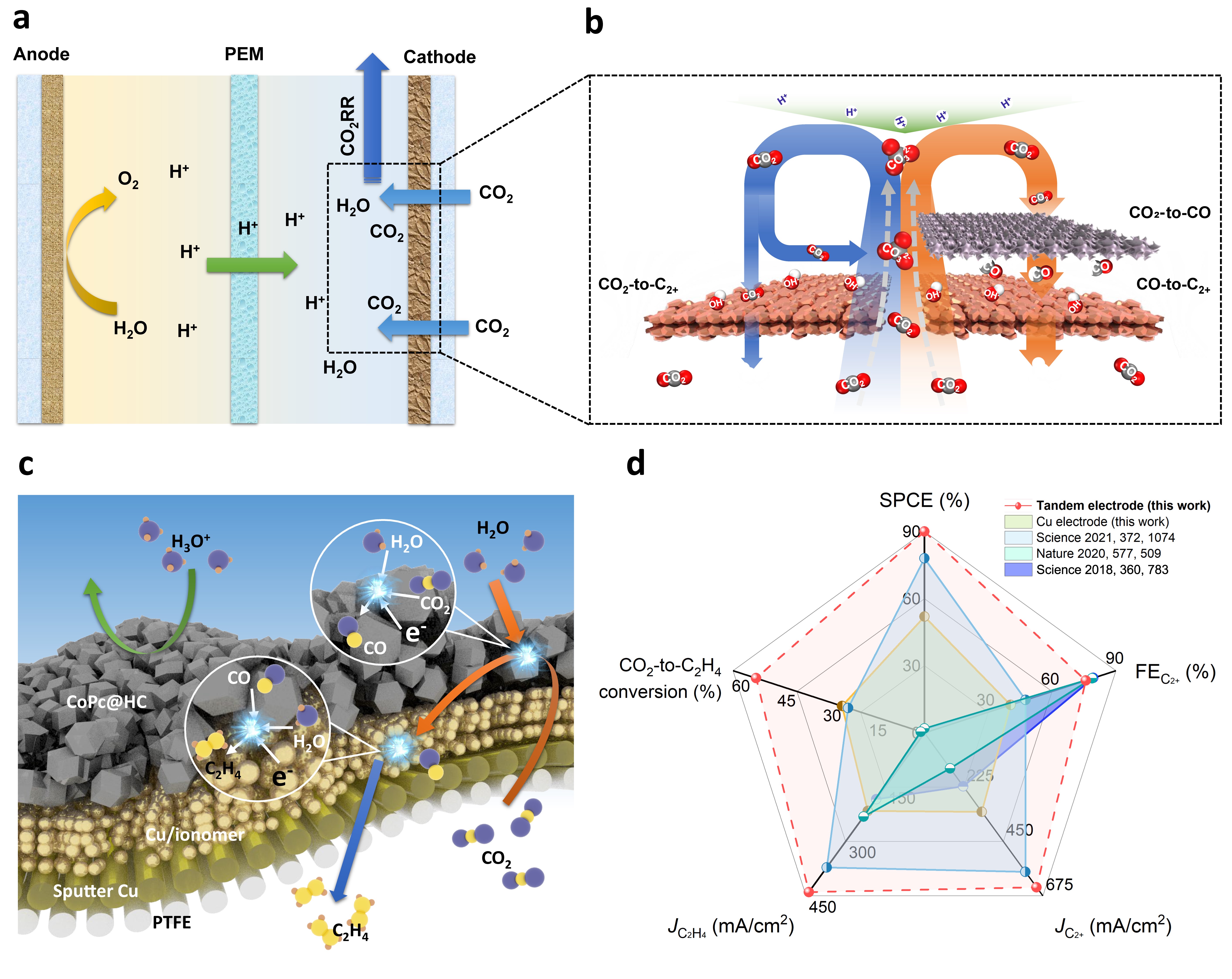
Figure 1. A tandem catalysis strategy for energy- and carbon-efficient CO2RR to multicarbon products. a, Ion transport and reactions in CO2RR in acidic media. b, Decoupling CO production and C-C coupling of acidic CO2RR via tandem catalysis. c, Electron transfer and mass transport in acidic CO2RR via tandem catalysis. d, Comparison of the SPCE, FE and partial current density of C2+ and ethylene as well as CO2-to-ethylene single-pass conversion of the tandem electrode with those of state-of-the-art electrodes (References: Science 2021, 372, 1074; Nature 2020, 577, 509; Science 2018, 360, 783).
Follow the Topic
-
Nature Nanotechnology

An interdisciplinary journal that publishes papers of the highest quality and significance in all areas of nanoscience and nanotechnology.


Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in