The missing intermediate to form ethanol from carbon dioxide
Published in Chemistry
The electroreduction of CO2 enables the conversion of carbon dioxide, water, and electric energy to a wide range of chemicals. So far, the scale up of this technology is limited to the production of carbon monoxide and formic acid, while generation of multi-carbon products is hindered by unstable activity and low selectivity.1 Beyond C1 molecules, many reaction products occur during CO2 electroreduction on copper, primarily ethylene, ethanol, and n-propanol, albeit up to 10 C2+ species have been reported in literature.2 Since pioneering reports by Hori,3 adsorbed carbon monoxide (*CO) has been indicated as a crucial intermediate along the C2+ products route. Besides, being ethanol and ethylene formation rates independent from electrolyte pH, the dimerization of CO via a single electron transfer is currently assumed as the rate-determining step for both these C2 products.4 After the formation of the dimer, several proton-coupled electron transfers take place till the formation of OCHCH2, the last common precursor between ethylene and ethanol. From this species, the final products originate. Due to the entanglement of the reaction pathways, the hydrocarbon and the alcohol usually appear together,2 limiting the selectivity of the process. Thus, the CO2 reduction puzzle seems resolved. However, while the CO dimer has been observed via spectroscopic techniques,5 there is no experimental evidence of the *OCHCH2 intermediate. Besides, remarkably, the ethanol-ethylene pair breaks during reduction of other precursors, such as glyoxal and glycolaldehyde for example.6
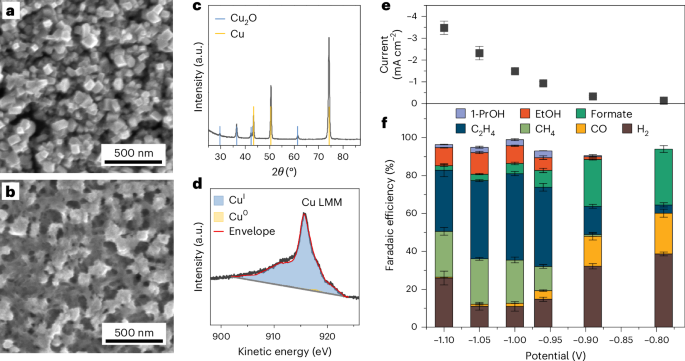
Here is where our story starts. Right after the completion of a previous study about CO coverage effects on ethylene selectivity,7 by analyzing new surface-enhanced Raman data during CO2 electroreduction, we bumped into four new vibrational signals, i.e., 1182, 1318, 1453, and 1595 cm–1. Surprisingly, these features occurred together at an applied potential between –0.85 V and –1.12 V vs RHE and were not fully compatible with the CO dimer. Were they spurious signals due to carbonate in solutions? We assessed perchlorate electrolytes, and we still observed analogous features. Did these vibrations involve carbon atoms? Our 13C/12C isotope exchange experiments confirmed, and analogous signals appeared during CO electroreduction. At that point, we could infer from literature that an intermediate with O and C atoms was responsible for the signals, yet we could not resolve its nature. We turned to density functional theory vibrational analysis. We computed the vibrational frequencies of seventeen potential adsorbates during CO2 electroreduction on nine copper active sites with different coordination numbers (data available in Supplementary Information). Just one molecule showed compatible signals, OCHCH2.
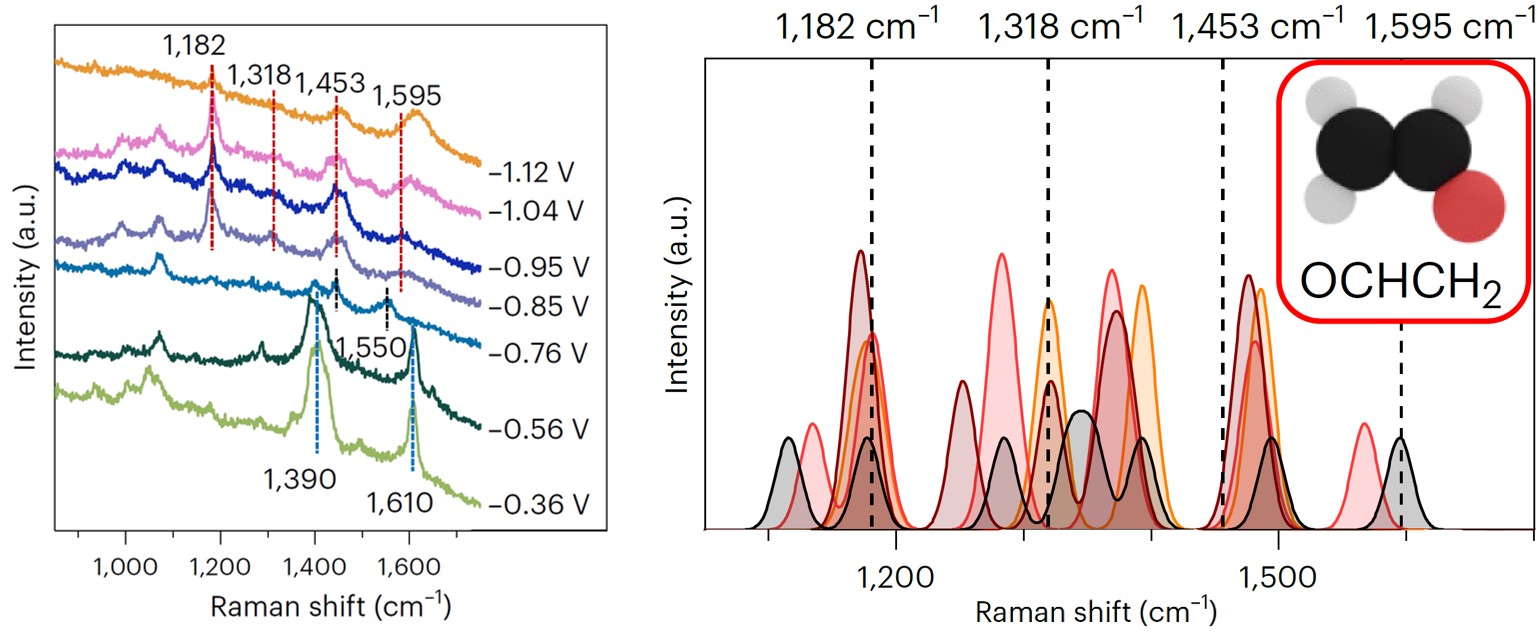
The first hypothesis was then straightforward. We had simply observed the OCHCH2 precursor of ethylene and ethanol, the missing tile of the CO2 electroreduction puzzle. To disregard this, however, the features were absent at –0.76 V vs RHE, where instead our online gas chromatograph already detected ethylene. There must have been a reason for OCHCH2 to appear later. We understood later that the reason was precisely called ethanol. In fact, we decided to run an ultimate benchmark test, i.e., to observe Raman signals from glyoxal reduction. Glyoxal (OCHOCH) selectively reduces to ethanol,6 and the simplest intermediate along the route is OCHCH2. Thus, if analogous signals were seen during glyoxal reduction, we could confirm OCHCH2 as ethanol precursor. We saw the signals. We had found the missing intermediate to form ethanol, but, even more importantly, we had disproved that this intermediate is an ethylene precursor, as generally assumed.4
Once understood its origin, we investigated why ethanol is the little brother of ethylene in terms of Faradaic efficiency. We first realized that, although ethanol selectivity was minor than ethylene, it had a steeper increase if a more negative potential was applied. Besides, we could demonstrate a higher abundance of undercoordinated defects within the ethanol selectivity region. Again, we turned to density functional theory to rationalize these insights. We assessed the binding energy of OCHCH2 on crystalline facets and undercoordinated distorted domains. Remarkably, the latter active sizes stabilized the intermediate way stronger than the crystalline ones. Such stabilizing effect was confirmed for adsorbed oxygen atoms, suggesting a general rule applicable to oxygenates. Besides, it was attributed to high compressive strain and deep s-band states.
Overall, the previous insights call for an updated C2+ reaction scheme. While the CO dimer is confirmed as the rate-determining step, ethylene and ethanol pathways overlap only till *CHCO. Then, a bifurcation occurs. Along the ethanol route, once the next intermediate forms, i.e., *CH2CO, it can either evolve to acetate, by desorbing and reacting with OH– in solution, or reduce to OCHCH2. From here, on distorted sites, acetaldehyde, ethanol, and 1-Propanol form as final products.
This fundamental work has three relevant implications. From the mechanistic point of view, ethanol and ethylene share a joint pathway only at preliminary reduction stages, till the *CHCO intermediate. From the experimental point of view, the observation of a new reaction intermediate highlights the progress in characterization techniques. From the catalytic point of view, by describing the active sites responsible for C2+ alcohols, we provide crucial guidelines to optimize their selectivity independently from ethylene’s one.
For more information, please read our Open Access article in Nature Energy at DOI: 10.1038/s41560-024-01633-4.
Density functional theory datasets can be freely accessed at the ioChem-BD8 database at DOI: 10.19061/iochem-bd-1-251.
References
- Shin, H., Hansen, K. U. & Jiao, F. Techno-economic assessment of low-temperature carbon dioxide electrolysis. Nat. Sustain. 4, 911–919 (2021).
- Kuhl, K. P., Cave, E. R., Abram, D. N. & Jaramillo, T. F. New insights into the electrochemical reduction of carbon dioxide on metallic copper surfaces. Energy Environ. Sci. 5, 7050–7059 (2012).
- Hori, Y., Takahashi, R., Yoshinami, Y. & Murata, A. Electrochemical reduction of CO at a copper electrode. J Phys Chem B 101, 7075–7081 (1997).
- Kortlever, R., Shen, J., Schouten, K. J. P., Calle-Vallejo, F. & Koper, M. T. M. Catalysts and reaction pathways for the electrochemical reduction of carbon dioxide. Journal of Physical Chemistry Letters 6, 4073–4082 (2015).
- Pérez-Gallent, E., Figueiredo, M. C., Calle-Vallejo, F. & Koper, M. T. M. Spectroscopic observation of a hydrogenated CO dimer intermediate during CO reduction on Cu(100) electrodes. Angew. Chem. Int. Ed. 56, 3621–3624 (2017).
-
Schouten, K. J. P., van der Ham, C. J. M., Qin, Z., & Koper, M. T. M. A new mechanism for the selectivity to C1 and C2 species in the electrochemical reduction of carbon dioxide on copper electrodes. Chem. Sci. 2, 1902 (2011).
- Zhan, C. et al. Revealing the CO coverage-driven C−C coupling mechanism for electrochemical CO2 reduction on Cu2O nanocubes via Operando Raman spectroscopy. ACS Catal. 11, 7694–7701 (2021).
- Álvarez-Moreno, M. et al. Managing the computational chemistry big data problem: The ioChem-BD platform. J Chem Inf Model 55, 95–103 (2015).
Follow the Topic
-
Nature Energy
Publishing monthly, this journal is dedicated to exploring all aspects of this on-going discussion, from the generation and storage of energy, to its distribution and management, the needs and demands of the different actors, and the impacts that energy technologies and policies have on societies.




Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in