Time will tell - CoraFluors, versatile TR-FRET probes for Chemical Biology
Published in Chemistry

Proximity drives biology. As chemical biologists, we are often interested how biomolecules interact with one another or with small molecule ligands. But, logistically speaking, where do you start when looking to profile the interaction of a small molecule with its hypothesized target(s)? There are many assay platforms that can be used to characterize the binding of small molecules (or other biopolymers) to proteins, including isothermal calorimetry (ITC), fluorescence polarization (FP), surface plasmon resonance (SPR), and biolayer interferometry (BLI). However, most of these techniques require isolation of the desired biomolecule, are low throughput and are unsuitable for analyzing the target in the complex environment present in either cell extract or within live cells.
The recent development of small, stable luciferase enzymes (e.g. NanoLuc)1 has provided a means to study proximity in cellular contexts via bioluminescence resonance energy transfer (BRET)2. BRET represents a unique flavor of assay methodologies that are founded on a physical phenomenon called Förster resonance energy transfer (FRET, Fig. 1a), where a donor fluorophore (or for BRET a bioluminescence-based donor) can non-radiatively transfer energy to an acceptor fluorophore when in close proximity (~5-10 nm, or approximately the size of a nucleosome)3. The acceptor emission can then be measured by a photodetector; these experiments are often run so that a plate reader (i.e. 384-well plate) or fluorescence microscope is the readout.
BRET-based techniques represent some of the most robust methods to study biomolecular interactions in both cell extracts and live cells where other methods have failed. Specifically, employing luciferases as fusion protein constructs in live cells has the advantages of allowing users to genetically encode a signal-generating enzyme and not requiring an excitation light source. While undoubtedly powerful, there are certain limitations of BRET-based strategies, including the continuous need for luciferase substrate to generate signal, and the lack of spatial and temporal control. Furthermore, since luciferases are enzymes, their applicability is confined to conditions that are compatible with their catalytic function (e.g. this complicates low-temperature, dynamic pH, and anaerobic measurements).
In recent years, the combination of time-resolved (TR) readouts with FRET-based applications has given rise to highly sensitive assay platforms (Fig. 1b). In particular, lanthanide complexes, most commonly of terbium (Tb) or europium (Eu), which possess luminescence lifetimes ranging from a few-hundred microseconds to milliseconds, are attractive probes for this approach4. While there has been a substantial number of luminescent lanthanide complexes reported, very few satisfy the requirements of being highly stable to biological media, having exceptional photophysical properties, and being synthetically readily accessible. The few lanthanide complexes that do meet these requirements can cause sticker shock – with some complexes retailing for upwards of $500 USD per microgram! This “minor” detail puts TR-FRET applications out-of-reach for many academic labs.
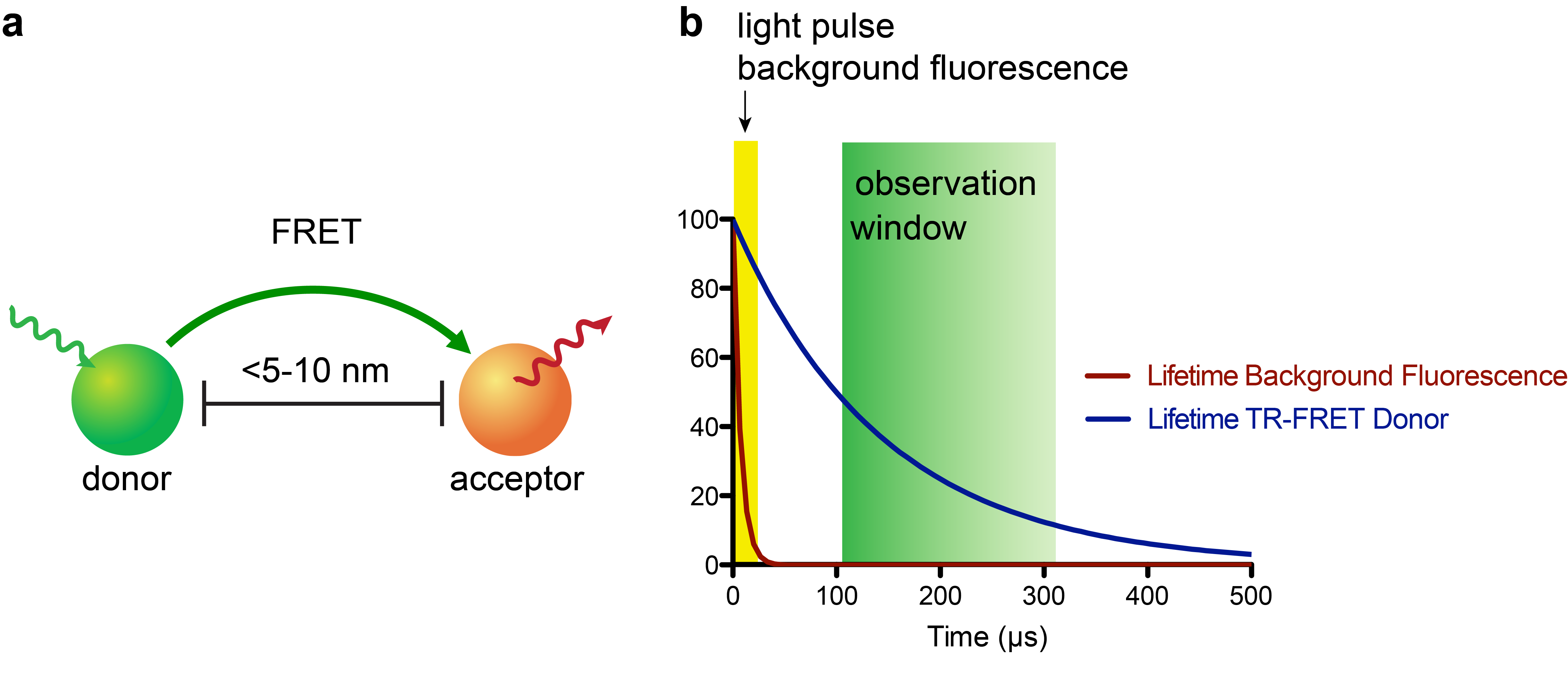
While TR-FRET synergistically combines high sensitivity with exquisite specificity and has found widespread use in cell-free assays, its use in live-cell platforms for intracellular targets has been challenging. The reason for this is that most lanthanide-based TR-FRET donors have poor cellular uptake, most likely because these probes are too polar to sneak through the lipid bilayer.
Our lab is interested in a number of different protein families. In particular, we are interested in exploring the biology of epigenetic regulators such as histone deacetylases (HDACs)5 and, more recently, the cellular redox regulator Keap1 and the targeting of which by reversible covalent small-molecule inhibitors6. We desired to develop a target-agnostic, robust, and sensitive assay platform that would allow us to study these protein targets in progressively more complex environments: first with isolated, recombinant proteins, then in cell extracts, and finally in living mammalian cells. The strengths offered by TR-FRET were obvious, but it was also clear that we needed to develop our own toolbox of high-performance TR-FRET probes that were synthetically accessible on a scale that would allow us to conjugate them to small molecules – along with being cell-permeable.
To this end, over the course of two years, my colleague and co-author Dr. Alena Kalyakina and I refined and optimized a versatile, scalable route to TR-FRET donors that we now call CoraFluors (Fig. 2a). The probes can be accessed in just 11 overall steps (>100 mg scale) and can be readily derivatized, here with halogen atoms (Cl, Br) to increase lipophilicity (membrane permeability) while retaining good photophysical properties (Fig. 2b).

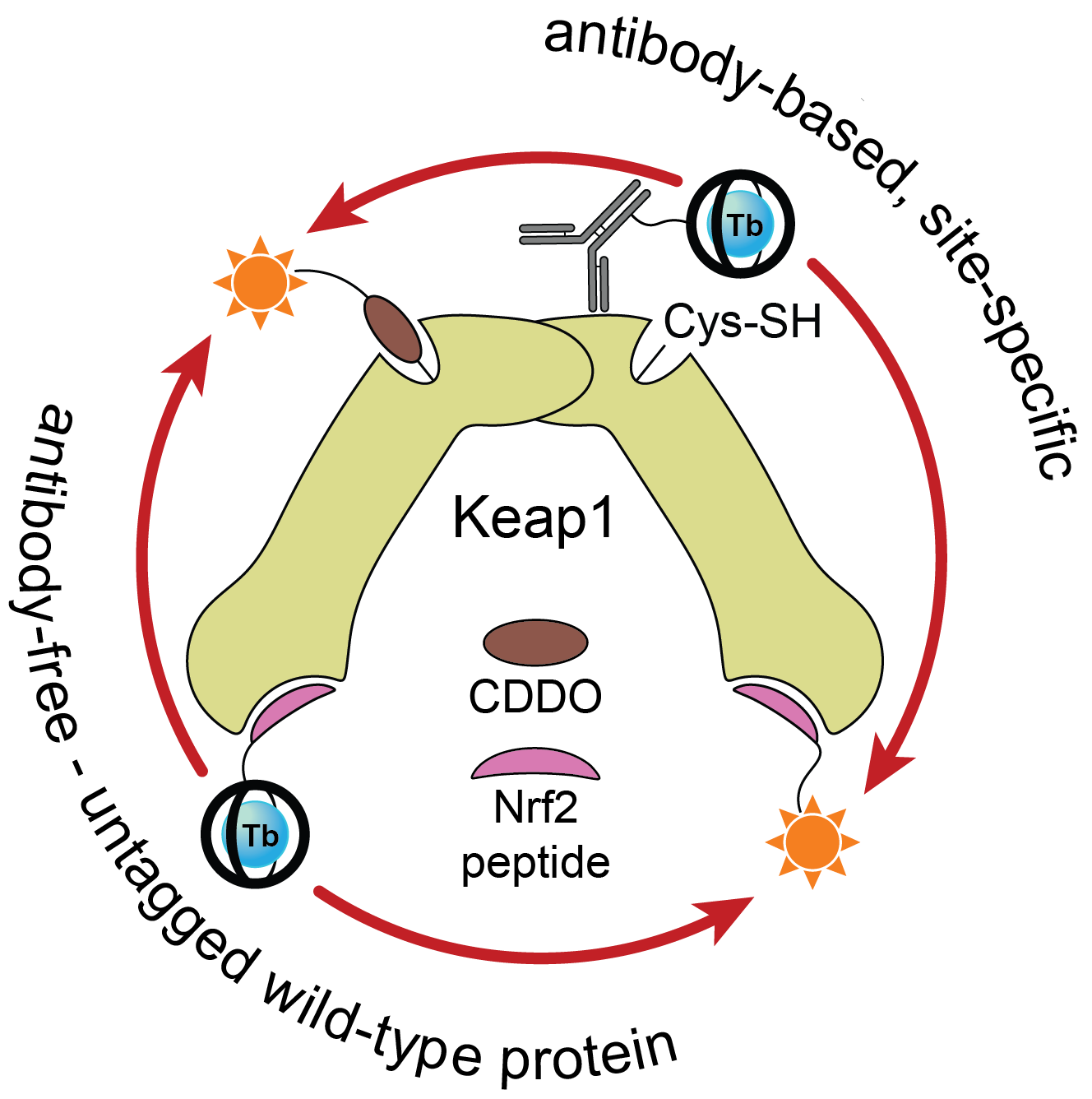
Once we had access to chemistry-enabling quantities of the CoraFluors, we conjugated them to small molecule probes, specifically with the goal of constructing a multimodal Keap1 assay platform. Our lab became interested in Keap1 when we co-discovered a mechanism of action of the reversible covalent inhibitor obtusaquinone, which causes degradation of Keap1 in cells and activation of the antioxidant response element pathway7. Using our functionalized probes, we developed a suite of assays compatible with full-length, wildtype protein and which do not require epitope-tagged antibodies to install the TR-FRET donor or acceptor (Fig. 3). These assay systems enable us to quantify the binding affinities of our test compounds, even allowing us to profile the kinetics of binding of reversible covalent inhibitors.
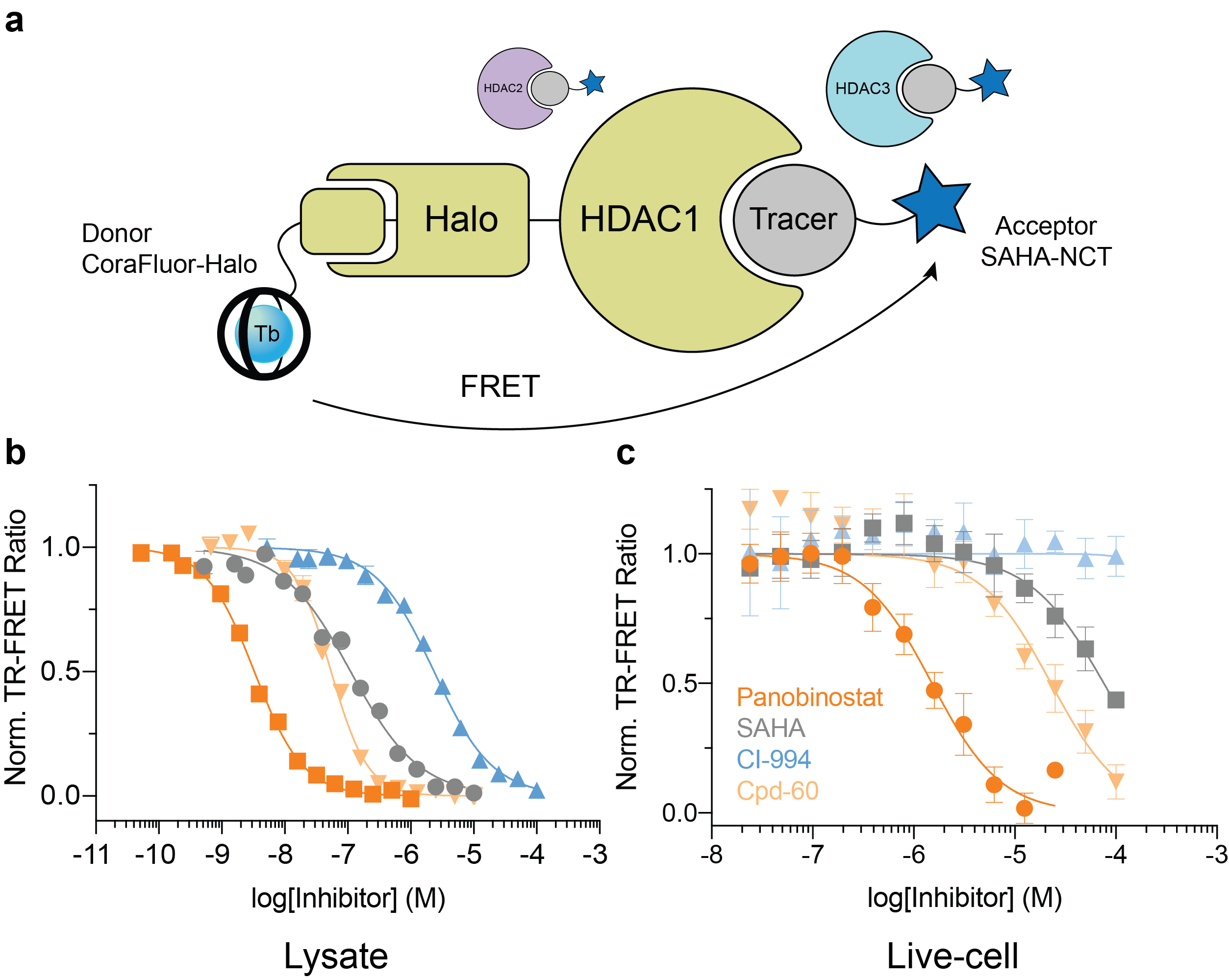
The final goal was to enable TR-FRET assay systems that were compatible with intracellular protein targets within live cells. To accomplish this feat, we employed the self-labeling HaloTag system8, in combination with HaloTag ligand-functionalized CoraFluors, to develop a TR-FRET platform to study target engagement of small molecule HDAC inhibitors (Fig. 4). The specific nature of the HaloTag renders the system compatible with cell lysates, where binding to a specific HDAC isoform can be monitored in the direct presence of all 11 mammalian isoforms (Fig. 4a). Extending this platform into cells, where HDAC1 resides in the nucleus, we used our cell-permeable CoraFluor-2-Halo complex in combination with an acceptor fluorophore-modified HDAC tracer (SAHA-NCT) to quantitatively measure inhibitor binding to HDAC1 in live HEK293T cells (Fig. 4b). Adjusting our observed EC50 values by accounting for ligand and tracer concentrations9 yielded apparent KD (KD,app) values that closely matched those reported in the literature and from our own cell-free HDAC profiling platform.
Circling back to the big picture, overall this work was motivated by our lab's desire to develop a reliable, flexible and target-agnostic assay platform capable of accurately and quantitatively profiling the binding kinetics and thermodynamics of the interactions between biomolecules with each other and with small molecules. Importantly, we needed to be able to measure target engagement in increasingly complex systems: from purified proteins to live cells. We believe that the CoraFluor TR-FRET probes described here along with some novel assay methodologies will provide chemical biologists with suitable opportunities to study their biological questions with exquisite versatility and assay performance.
For more information, please read our article in Nature Chemical Biology at https://www.nature.com/articles/s41589-021-00877-5
References
- Machleidt, T. et al. NanoBRET—A Novel BRET Platform for the Analysis of Protein–Protein Interactions. ACS Chemical Biology 10, 1797-1804 (2015).
- Robers, M.B. et al. Target engagement and drug residence time can be observed in living cells with BRET. Nature Communications 6, 10091 (2015).
- Algar, W.R., Hildebrandt, N., Vogel, S.S. & Medintz, I.L. FRET as a biomolecular research tool — understanding its potential while avoiding pitfalls. Nature Methods 16, 815-829 (2019).
- Bünzli, J.-C.G. Lanthanide Luminescence for Biomedical Analyses and Imaging. Chemical Reviews 110, 2729-2755 (2010).
- Bradner, J.E. et al. Chemical phylogenetics of histone deacetylases. Nature Chemical Biology 6, 238 (2010).
- Cuadrado, A. et al. Therapeutic targeting of the NRF2 and KEAP1 partnership in chronic diseases. Nature Reviews Drug Discovery 18, 295-317 (2019).
- Badr, C.E. et al. Obtusaquinone: A Cysteine-Modifying Compound That Targets Keap1 for Degradation. ACS Chemical Biology (2020).
- Los, G.V. et al. HaloTag: A Novel Protein Labeling Technology for Cell Imaging and Protein Analysis. ACS Chemical Biology 3, 373-382 (2008).
- Yung-Chi, C. & Prusoff, W.H. Relationship between the inhibition constant (Ki) and the concentration of inhibitor which causes 50 per cent inhibition (I50) of an enzymatic reaction. Biochemical Pharmacology 22, 3099-3108 (1973).
Follow the Topic
-
Nature Chemical Biology
An international monthly journal that provides a high-visibility forum for the chemical biology community, combining the scientific ideas and approaches of chemistry, biology and allied disciplines to understand and manipulate biological systems with molecular precision.



Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in