Tree population genomics. A story of 10 years of research and 10 million years of evolution
Published in Ecology & Evolution, Sustainability, and Genetics & Genomics
It was 2015 and I was on parental leave with my second child when I got a phone call from my former supervisors and colleagues asking if I was interested in joining a European Union grant proposal on forest tree genetic diversity. Of course, I said yes without really knowing beyond that there would be an opportunity to work with the European forest genetic community (talking about baby brain).
Now it is October 2024, my daughter will turn 10 soon, and the comparative genomic analysis is finally published. What happened in between?
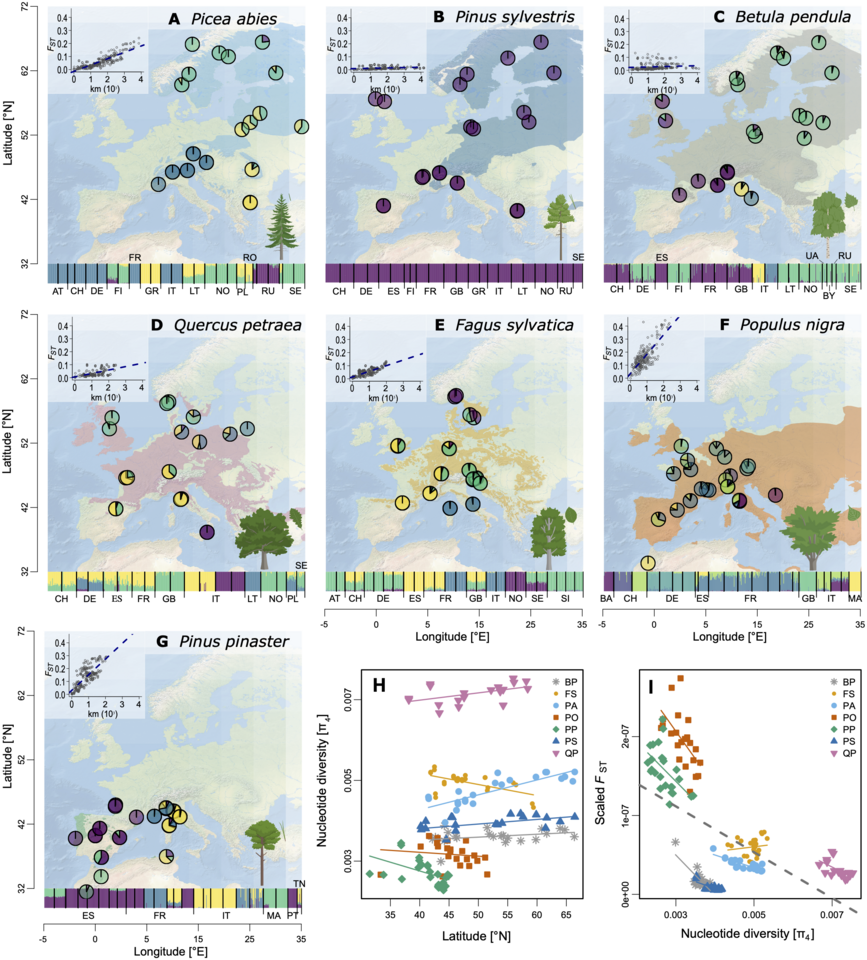
The project GenTree was funded by the European Union’s Horizon 2020 research and innovation programme and began in 2016 with 22 partner organizations from 14 countries all coordinated by French INRA. As one of the goals of the project was to quantify key demographic processes affecting forest genetic diversity in Europe, we had to do extensive sampling across many countries and coordinate tissue collection, DNA extractions and DNA sequencing. Thanks to the project partners and efficient collaborations, the sampling eventually covered the European part of several species’ distributions. Even though we were splitting the work, our southern European partners conducted the majority of the work simply due to higher tree species abundance towards more southern latitudes. The population genetic analysis focused on seven species (Scots pine, Norway spruce, maritime pine, silver birch, black poplar, European beech, pedunculate oak), but even more sampling was done for phenotypic and environmental data. The same material was also used to establish a dendroecological and leaf variation datasets.
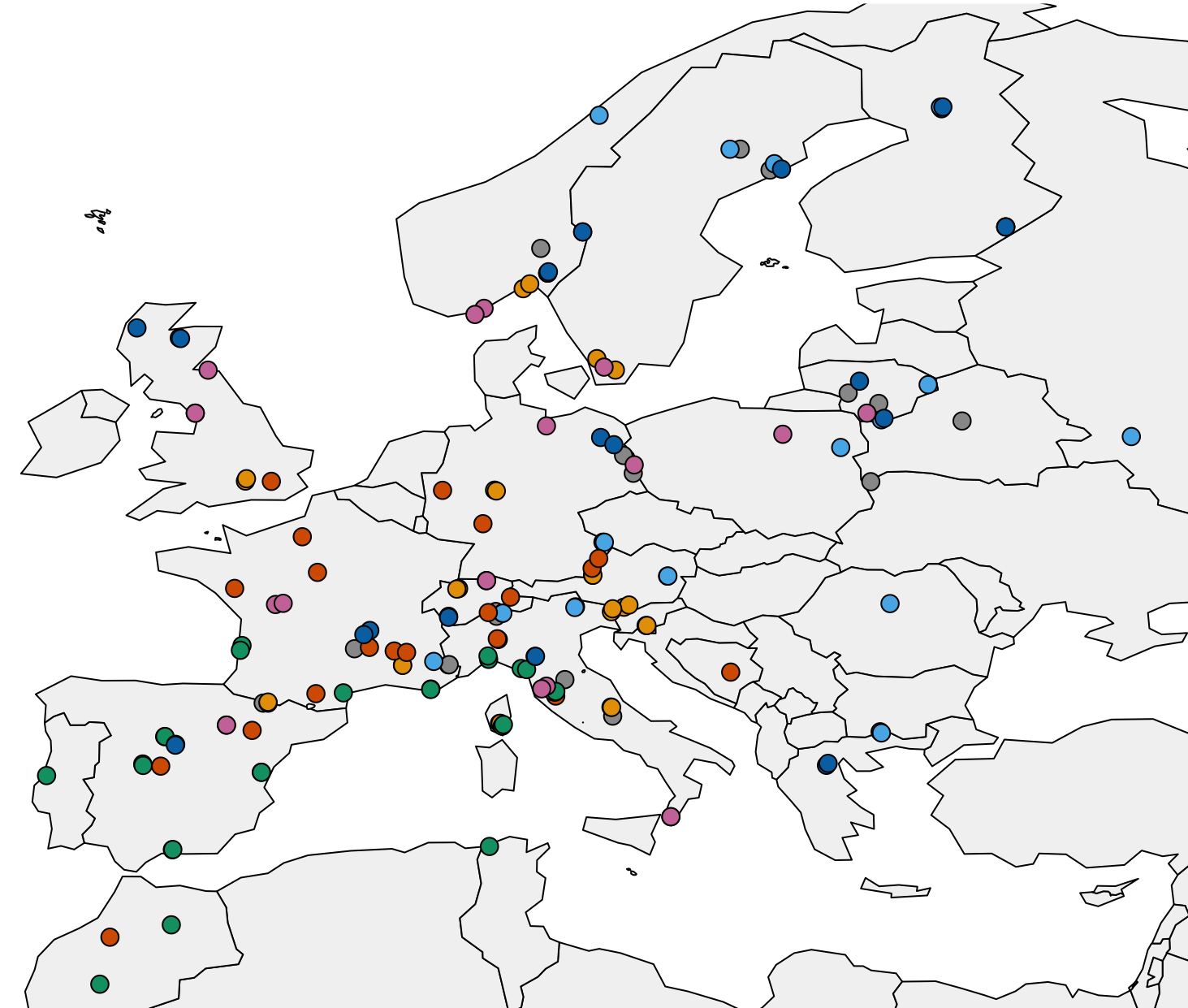
Sampling locations
As we were studying both angiosperm and gymnosperm trees’ genetic diversity in a comparative way, the next challenge was to decide how to measure genetic diversity as harmoniously as possible. Gymnosperm genomes are very large and repetitive, and the budget limited, thus the whole genome sequencing of all 3,407 study trees was not an option. As an elegant and cost-effective compromise, we chose to use targeted sequencing of some shared orthologous genes in functions of interest, some species-specific genes, and some randomly selected genes.
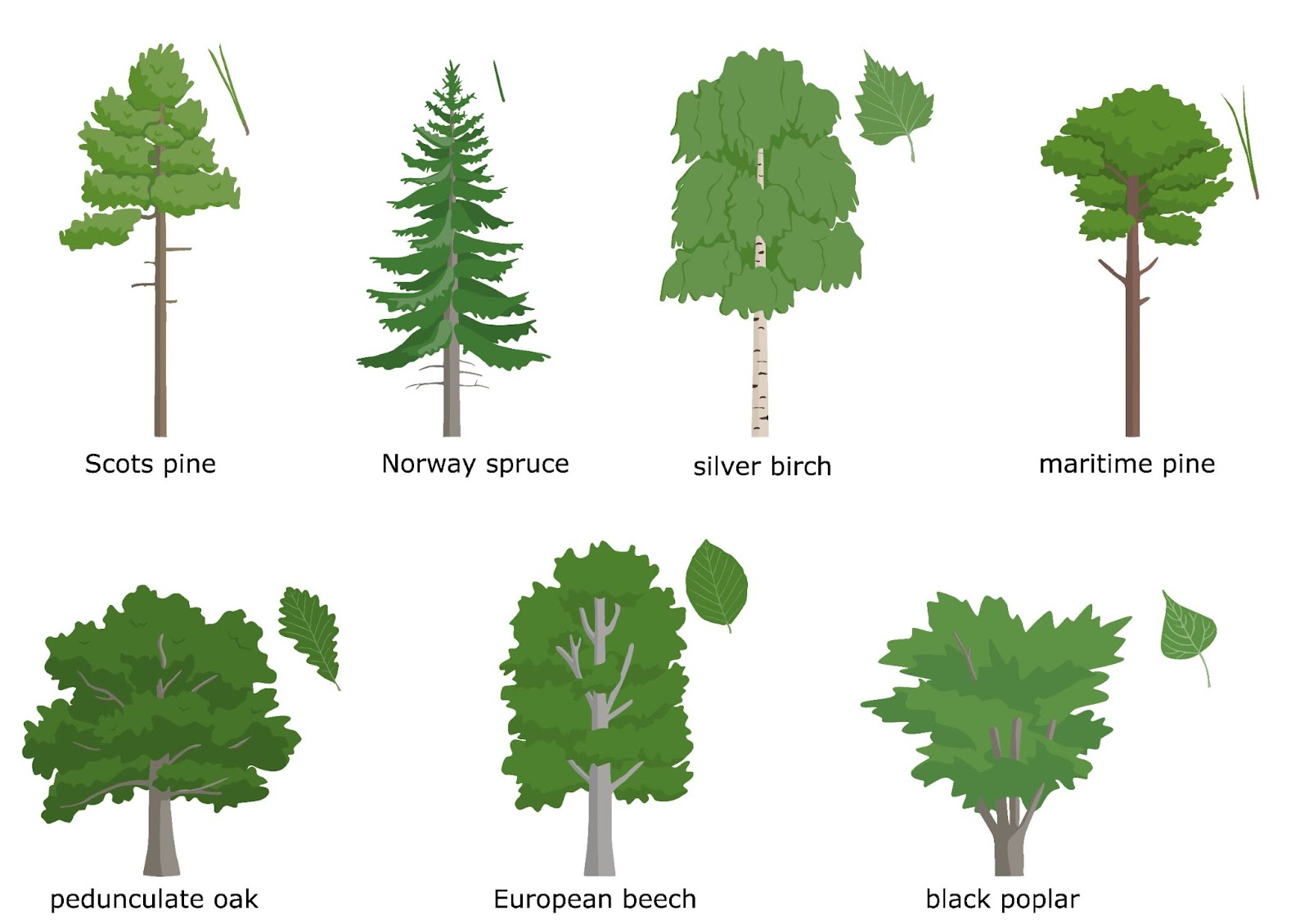
The studied species
Little by little, DNA sequence data started to accumulate, and we had to make countless decisions on data curation and bioinformatic analyses. What data is deemed too poor to include? Which reference genomes to use for mapping? How do we identify sampling mistakes (wrong species, duplicates, clones)? What do we do with weird samples that are potentially hybrids or highly admixed individuals between two species? Also, a significant amount of time was spent on harmonizing the sample labeling and naming! Every species was a little bit different with its own issues and we needed the expertise of the whole group to address each of them. For example, Populus has clonality and some very common cultivars that needed to be excluded, and specific oak and birch species can be hard to identify. Some species did not have a reference genome and we had to decide which reference to use. We even tried to make a de novo assembly of the targeted regions for species without a reference genome. To make it all work, good communication was essential! I was leading the population genetic analysis tasks and this was my first time in such a role, and I had to put all my organizational and communication skills to use and learn some new ones along the way to make it work.
Eventually, we were happy with the data and ready for the interesting part: estimation of genetic diversity parameters and inferring population demographic history. We chose to use two inference methods that are based on the site-frequency-spectrum of alleles, Stairway Plot 2 and fastsimcoal2, testing different sampling levels to control for the sampling and population structure effect on the inference. In the big picture, different methods and sampling strategies were congruent with each other.
Demographic inference methods yield estimates of effective population size (Ne) and timing of divergence among populations. The scaling into years depends on estimates of mutation rate and generation time. The Ne itself is a rather complicated parameter, but in simple terms it describes the rate of genetic lineages finding their most common recent ancestor at the given time point. In times of high Ne, the rate is low and during low Ne, the rate is high. Although it is not completely equivalent to the actual census population size, it is a useful parameter that summarizes the historical population genetic factors that have affected the genetic diversity observed in the data today.
In all forest tree species we studied, a major part of genetic diversity originated from hundreds of thousands or even millions (e.g., in oaks) of years ago. It is fascinating to think that the genetic polymorphisms we observe today have already emerged a million years ago and have been segregating in the population ever since! Another interesting observation was that the main genetic groups within species have already diverged by 0.6 Mya to 17 Mya. Thus, the common interpretation of forest tree genetic structure being a consequence of separation between populations during the Last Glacial Maximum (20 kya) is highly unlikely.
Surprisingly to us, all species also had an indication of Ne growth during the past glacial cycles, and the Ne changes did not seem to correspond with glacial and interglacial periods. The species that we studied are both long-lived and common and have a proven ability to quickly adapt to new environmental conditions. They also have very efficient gene flow. These characteristics probably allow tree populations to behave as one large metapopulation connected by gene flow that always thrives somewhere in large enough populations to maintain or even accumulate genetic diversity. The trees just live in different time scales and stick together, like the ents in Tolkien’s The Lord of the Rings!
The project funding ended years ago, and I am very grateful to all my colleagues, especially at Uppsala University, WSL Switzerland, and University of Helsinki who found the work interesting and important enough to push it through on top of their current projects for several years. I am also very proud that all the GenTree datasets are publicly available for anyone to use!
Follow the Topic
-
Nature Communications

An open access, multidisciplinary journal dedicated to publishing high-quality research in all areas of the biological, health, physical, chemical and Earth sciences.
Related Collections
With Collections, you can get published faster and increase your visibility.
Women's Health
Publishing Model: Hybrid
Deadline: Ongoing
Biosensing
Publishing Model: Hybrid
Deadline: Jun 30, 2026



Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in