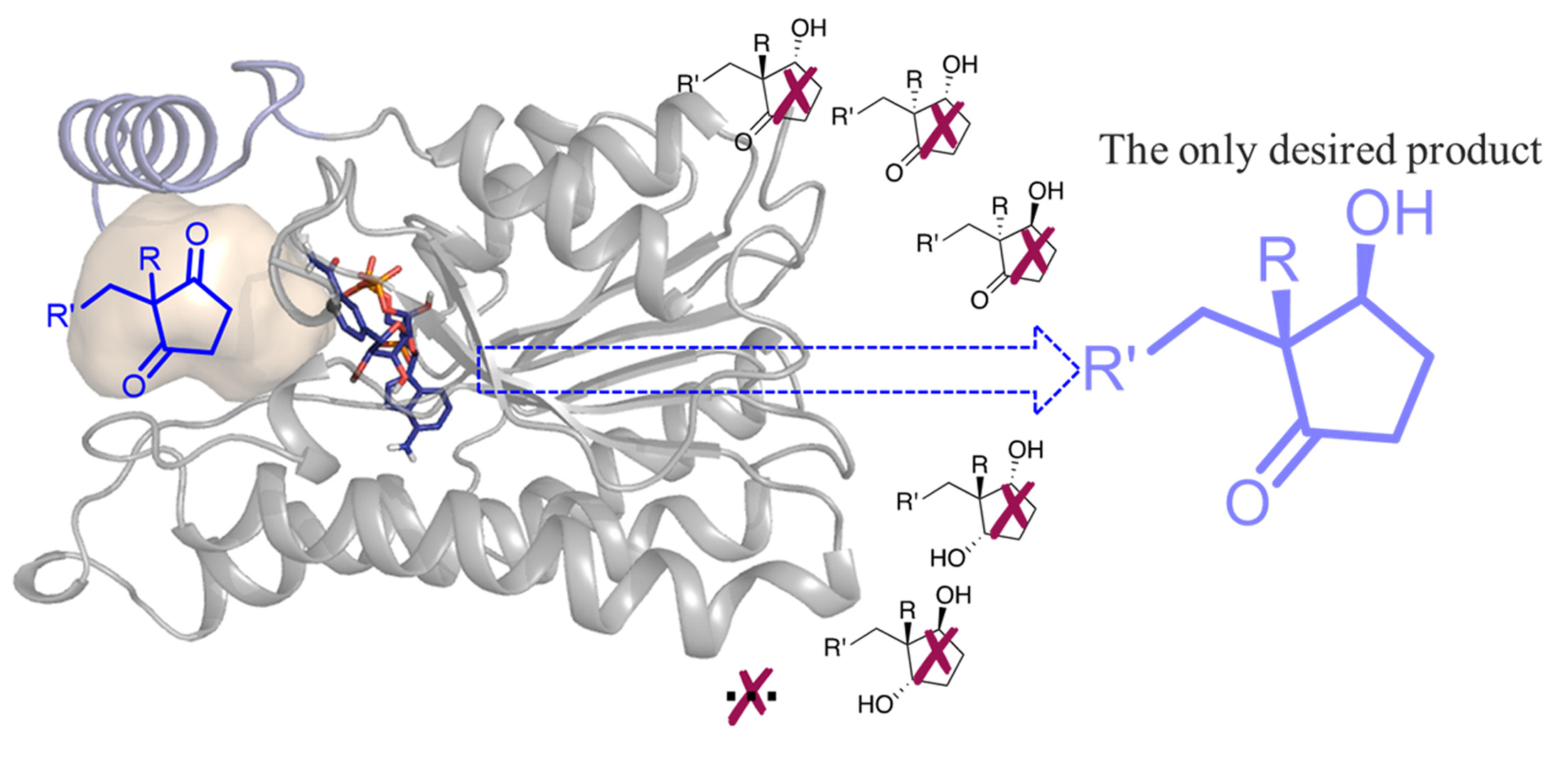
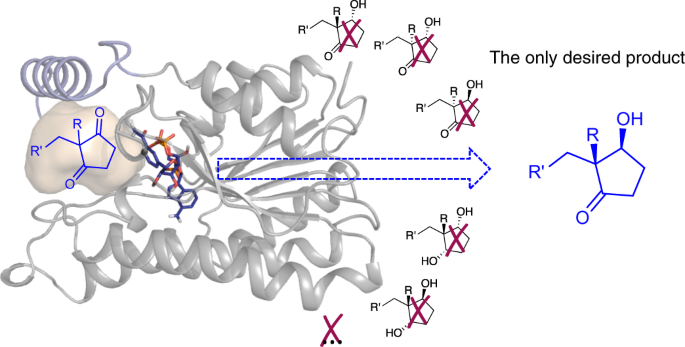
Ethyl secol is the key intermediate for the preparation of the steroidal drugs, gestodene and levonorgestrel, which are among the top 200 pharmaceutical products by retail sales in 2018. Reductive desymmetrization of ethyl secodione is a straightforward way to obtain the single stereoisomer of ethyl secol (Fig. 1).
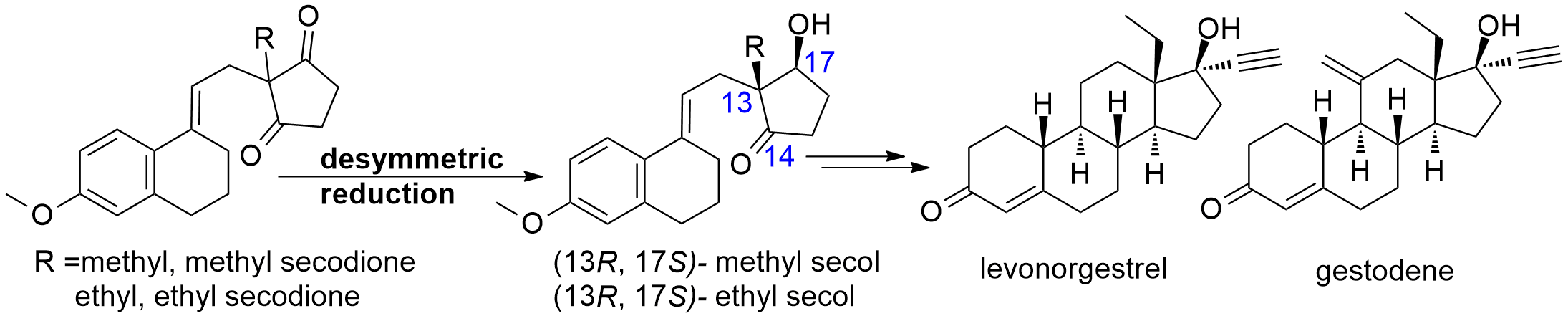
Fig. 1 Desymmetric reduction of methyl or ethyl secodione as the key step in the production of steroidal drugs
Based on the character of substrate, without excellent regio-, stereoselectivity, ethyl secol (one carbonyl group is reduced to form two chiral centers) and ethyl secodiol (two carbonyl groups are reduced to form three chiral centers) are the possible products. As so far, efficient chemical or biocatalytic reduction methods have been barely reported. Our research group has been working on enzymatic reduction for a while, so we became interested in finding enzyme catalysts for this highly challenging reductive desymmetrization.
As a start of this project, Hongliu and I screened nearly 100 carbonyl reductases available in our laboratory using ethyl secodione as the substrate. The results showed that none of the carbonyl reductases gives enough conversion and good stereoselectivity, suggesting the difficulty of the desymmetric reduction. We then performed structure-guided directed evolution on the carbonyl reductase from Ralstonia sp. (RasADH). The substrate-enzyme docking analysis suggested that F205 blocks the substrate entry and binding, so we first did saturation mutagenesis and screening of this amino acid residue. Mutant F205A was found to improve the enzyme activity about 18 fold. Further three rounds of mutation and screening gave a mutant F12 with 183-fold enhanced enzyme activity and perfect stereoselectivity (Fig. 2). Mutant F12 showed high productivity not only for (13R, 17S)-ethyl secol (now the best catalyst, 20 g/L in 6 hours), but also for other (2R,3S)-2,2-disubstituted-3-hydroxycycloketones.
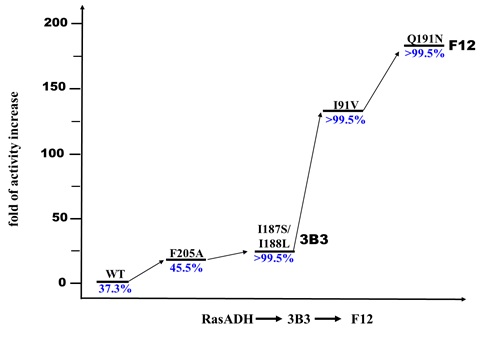
Fig. 2 The iterative stepwise improvement process of RasADH directed evolution
It is worthy noting that a new screening method was adopted in our study. In order to improve the screening efficiency, we used the alcohol product as the screening substrate for the oxidation reaction. Furthermore, the desired stereoisomer and the mixture of the unwanted stereoisomers were used as the substrate to screen the mutant library separately by measuring the absorbance change of the cofactor. In this way, the activity and stereoselectivity of the hits can be determined at same time, avoiding the normally used chiral HPLC or GC analysis for stereoselectivity screening.
In order to understand how mutant F12 shows strikingly increased activity and stereoselectivity, we went on to get the crystal structure of F12. Fortunately, Hongliu got the crystals of F12 complexed with NADPH and (13R, 17S)-ethyl secol, and Weidong collected the X-ray refraction data and solved the crystal structures. In the summer of 2018, Dunming met Sílvia and Miguel Angel at Biocatalysis Gordon Research Conference in Biddeford and discussed the collaboration on this project. Miguel Angel computationally explored the conformational dynamics of the wild-type and F12 variant, which revealed drastic differences in the flexibility of a lid region of the enzyme key for activity improvement and stereoselectivity control mechanism.
Up next: This just the start of the exciting project on the enzymatic desymmetric reduction of prochiral cyclic and acylic 1,3-diketones and effective construction of 3-hydroxyketones with multiple chiral centers. We are now focusing our studies on engineering enzymes for synthesizing (2R,3R)-, (2S,3S)-, (2S,3R)-2,2-disubstituted-3-hydroxycycloketones, and extending the desymmetric reduction to other cyclic and acylic 1,3-diketones.
Follow the Topic
-
Nature Catalysis
This journal brings together researchers from across all chemistry and related fields, publishing work on homogeneous catalysis, heterogeneous catalysis, and biocatalysts, incorporating both fundamental and applied studies.


Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in