von Willebrand disease is more common than what was previously thought
Published in Genetics & Genomics
von Willebrand factor (VWF) is a large plasma glycoprotein that is essential for normal hemostasis by mediating platelet adhesion to damaged vascular subendothelium and subsequently platelet aggregation and by protecting coagulation factor VIII (FVIII) from proteolysis. A deficiency or dysfunction of VWF leads to the most common inherited bleeding disorder, von Willebrand disease (VWD). VWD arises from two types of quantitative or qualitative VWF defects. The quantitative defect can be partial or complete leading to type 1 or type 3 VWD. VWF qualitative defects result in four different VWD types 2 (2A, 2B, 2M, and 2N). The genetic variants responsible for type 1 (mostly dominant) and 3 VWD (recessive) are spread across the 52 exons of the VWF gene (VWF), whereas type 2 VWD variants are confined to VWF functional domains.
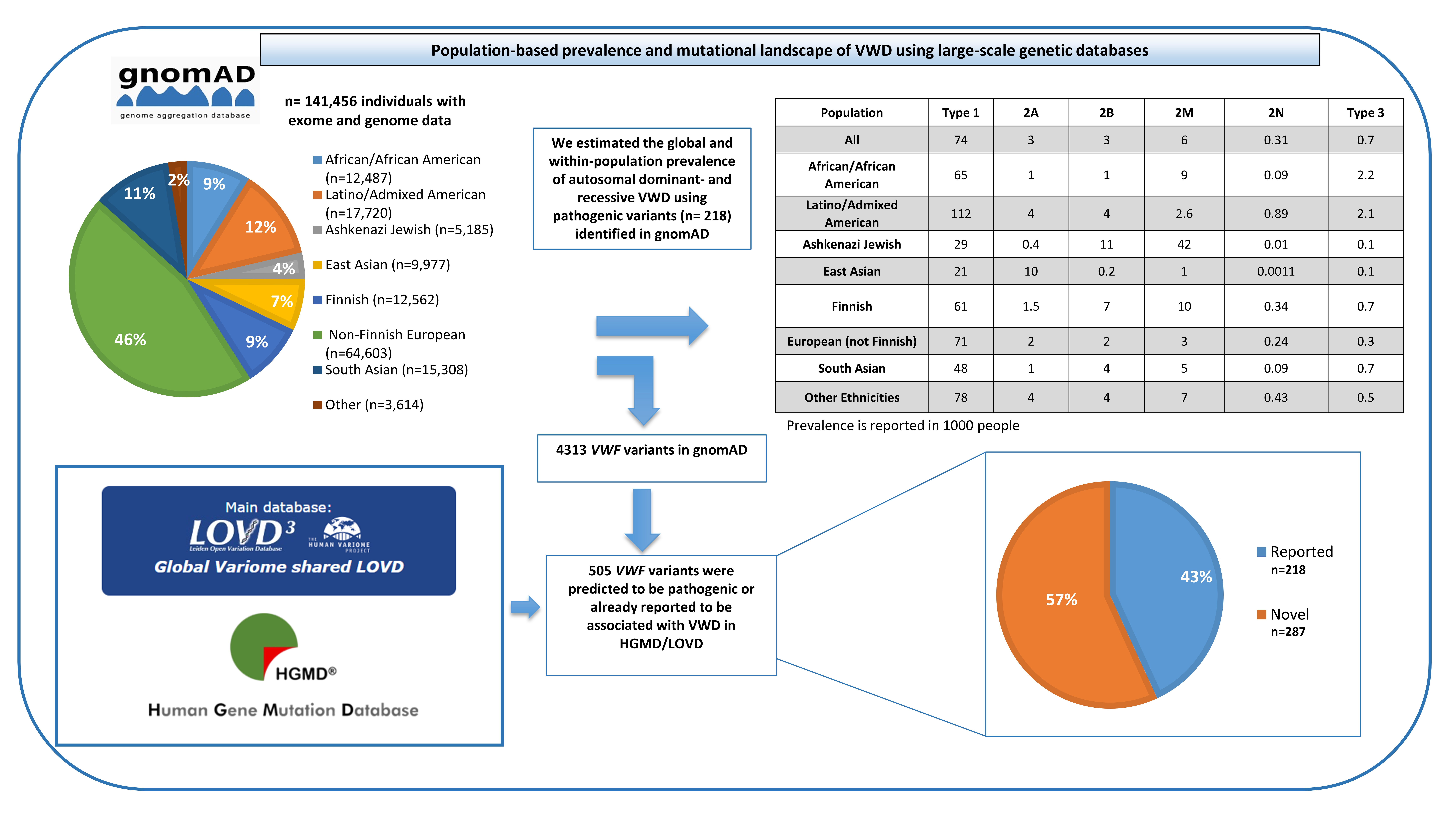
According to previous studies, VWD prevalence is estimated to vary between 0.6% and 1.3%, even though on the basis of cases referred to specialized centers about 1 case per 1,000 is estimated to have clinically relevant VWD. Despite this, the true prevalence of VWD has not been accurately established due to a lack of prospective and systematic studies and to the fact that some patients with VWF variants are asymptomatic or show mild clinical symptoms. In addition, the number of people investigated in the aforementioned studies was not large enough to estimate global VWD prevalence and these studies were limited to a small number of geographic areas. Nowadays, a growing number of large-scale population-based sequencing studies are being conducted using next-generation sequencing (NGS). By using genetic data and specialized statistical techniques (Hardy–Weinberg principle), the estimate of disease prevalence can be obtained by means of allele frequency information from a large number of sequenced samples. In this recent study entitled ”Population-based prevalence and mutational landscape of von Willebrand disease using large-scale genetic databases”, we chose to examine the global mutational landscape of VWF and to assess the worldwide and within-population prevalence of inherited VWD by analyzing exome and genome data of more than 141,000 individuals gathered by the genome Aggregation Database (gnomAD). We further extended and deepened data mining to the two primary databases containing VWF variants, i.e., the Leiden Open Variation Database (LOVD) and the Human Gene Mutation Database (HGMD) with the goal of analyzing the global mutational spectrum of VWD (Figure 1).
Findings
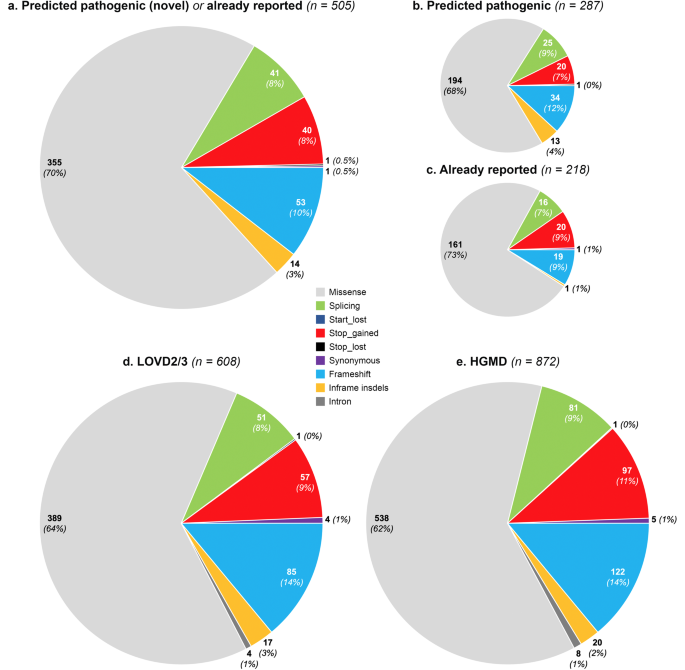
We collected high-quality data from gnomAD including 141,456 subjects with different ethnicities (Figure 1), including 12,487 Africans/African Americans, 17,720 Latinos/Admixed Americans, 5,185 Ashkenazi Jews, 9,977 East Asians, 12,562 Finnish Europeans, 64,603 non-Finnish Europeans, 15,308 South Asians and also 3,614 additional persons without an assigned ethnicity. A total of 4,313 different genetic variants were identified in the VWF in the gnomAD population. Following a conservative approach to classifying variants as pathogenic (i.e., as responsible for VWD), we found 505 distinct VWF deleterious variants of which 287 (57%) have not been previously reported to be associated with VWD, whereas 218 (43%) had been already reported. Out of these 505 selected pathogenic variants, 244 (48%) were unique, each variant being identified in one subject only. Among a total number of 282,912 alleles analyzed, 31,785 contained VWF pathogenic variants and only 2.9% of these affected alleles were carrying the novel variants. In the East Asian population, as many as 18.9% of affected alleles carried novel variants, whereas among other ethnicities the impact of novel variants was considerably lower (1.3%-4%). Among all pathogenic variants in gnomAD (n= 505) and also among those novel (n= 287), fewer variants were identified in the VWF D'-D3, A1-A2, and CK domains. However, more novel variants were identified in the D1-D2, A3, D4, and C1-C6 domains.
When data analysis was extended to the two main databases containing VWD-associated variants (HGMD and LOVD), 1024 different VWF variants were found to be associated with VWD so far, 927 of them being single nucleotide variant (SNV) and short insertions/deletions.
To calculate the true global prevalence of dominant (types 1, 2A, 2B, and 2M) and recessive (types 3 and 2N) VWD, we used only the variants reported to be associated with VWD in the gnomAD population (n= 218) with an already established autosomal dominant or recessive inheritance pattern. Accordingly, the global prevalence of dominant VWD in 1000 individuals was established to be 74 for type 1, 3 for 2A, 3 for 2B, and 6 for 2M. The global prevalences for recessive VWD forms (type 2N and type 3) were 0.31 and 0.7 in 1000 individuals, respectively (Figure 1). In addition, it appears that VWD prevalence differs among various populations. The high VWD prevalence established in this large-scale genetic epidemiology study indicates that the genetic predisposition to develop VWD due to VWF variants is likely to be more common than hitherto reported and also highlights that many patients carrying these variants are still not diagnosed. These data provide a hint that VWD is likely to be grossly underdiagnosed worldwide, which could contribute to undertreatment, significant (avoidable) morbidity, and health care system burden.
Follow the Topic
-
npj Genomic Medicine
This is an international, peer-reviewed journal dedicated to publishing the most important scientific advances in all aspects of genomics and its application in the practice of medicine.

Related Collections
With Collections, you can get published faster and increase your visibility.
Diploid Genome and Disease
Publishing Model: Open Access
Deadline: Feb 07, 2027
Artificial Intelligence in Genomic Medicine
Publishing Model: Open Access
Deadline: Sep 23, 2026




Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in