What does the nasal microbiome tell us about granulomatosis with polyangiitis?
Published in Microbiology
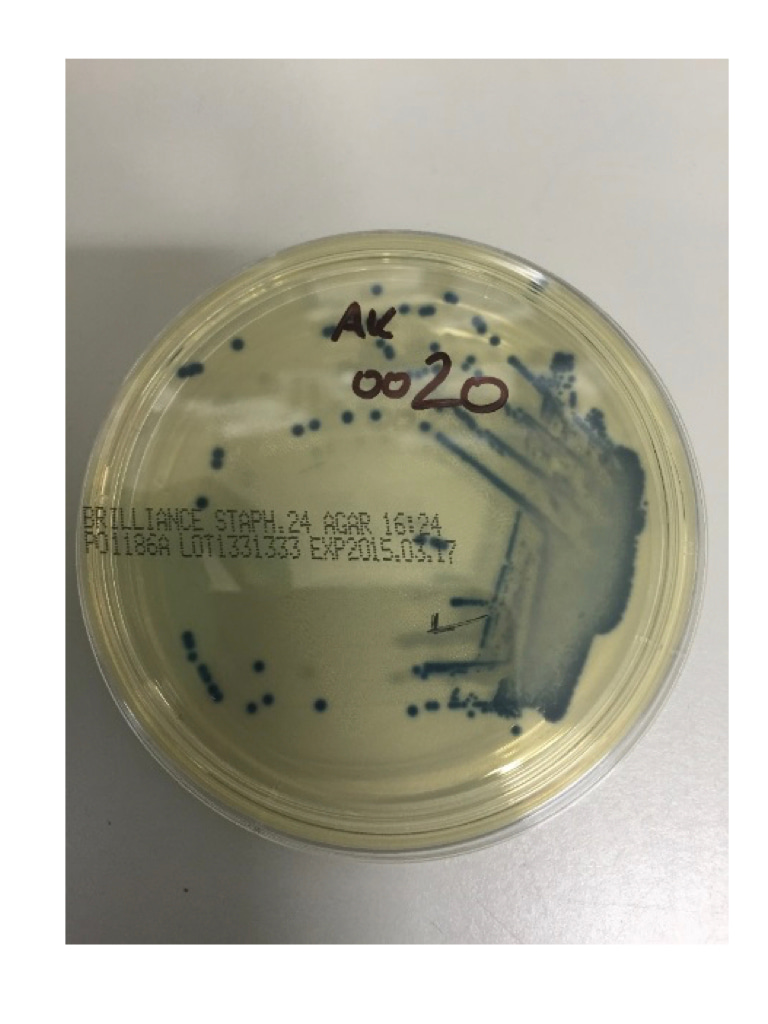

Granulomatosis with polyangiitis (GPA) is a multi-system autoimmune disorder. The exact pathogenesis is still unclear, but genetics and also environmental risk factors have been implicated. GPA is a relapsing-remitting disease and clinical predictors of disease relapse have been identified. Patients with GPA have a higher rate of nasal colonisation by Staphylococcus aureus (60-70%) than the general population (~25%), and the presence of persistent colonisation may be associated with an increased risk of disease relapse during follow-up. Recent studies have in part challenged this assumption. Thus, we designed a study to investigate the nasal microbiome and if changes are involved in disease pathogenesis or recurrence of disease or not.
In our study (link to our study) we characterised the nasal microbiome by bacterial 16S rRNA sequence analysis and in a particular depth for Staphylococcus species using shot gun metagenomic sequencing.
Since GPA represents a rare disease, numbers of patients recruited were limited to 12 with active disease and 44 with inactive GPA. As comparators we investigated the nasal microbiome in 13 disease controls, 11 unrelated healthy controls and 4 related healthy household controls. In addition, nasal swabs were used for culturing and 29 subjects (34.5%) were positive for S. aureus which were used for antimicrobial susceptibilty testing.
The bacterial 16S rRNA gene analysis identified three distinct microbial hierarchical clusters in the heatmap plot. Of these, one cluster was dominated by S. aureus and mainly contained patients with GPA. Statistical analysis using PERMANOVA test and NMDS plotting confirmed a significant difference beta diversity (between microbial cluster differences) between the GPA patients and the healthy control cluster. Antimicrobial susceptibilty testing revealed that none of the S. aureus isolates were methicillin resistant (MRSA), and three isolates were completely susceptible to all antibiotics testedWe then used shotgun metagenomic sequencing to obtain a greater resolution in terms of species present in the nasal microbiome. By shotgun sequencing we could retrieve 198 different Staphylococcus hits of which 38 hits could be classified at the species level and the other hits were mainly unclassified S. species (clones). Seven S. species were in the top 1% abundance group. Statistical analysis of the shot gun sequenced Staphylococcus species revealed that the active GPA patients were different from the healthy and disease controls.
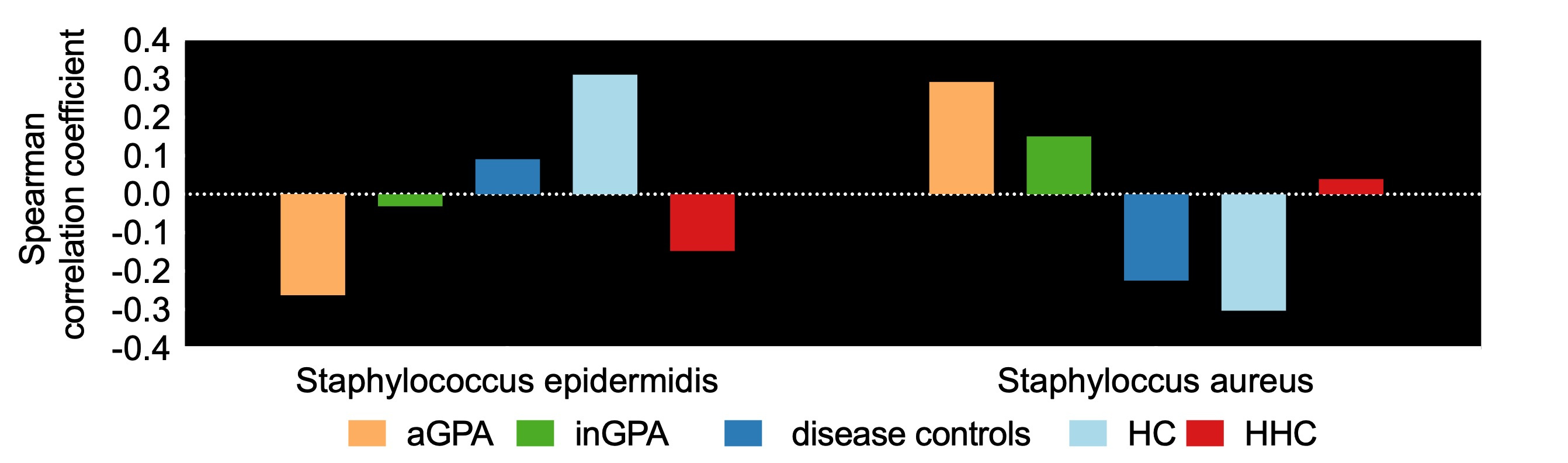
Differences of the two most abundant shotgun sequenced Staphylococcus species among the five different sample groups were found, as identified by shotgun sequencing.
Interestingly, we observed presence of S. pseudintermedius in patients and controls, which has not been described in this frequency before. S. pseudintermedius is a commensal and opportunistic pathogen of dogs and cats, that frequently causes soft tissue and skin infections and is increasingly recognized as zoonosis in humans (1). If S. pseudintermedius has a role in the pathology of GPA disease needs to be determined.
We next moved from the resolution of bacterial species to the level of the gene. We performed functional profiling by utilising shotgun sequenced data to investigate if the abundance of genes, and genes that are part of the same pathways were differentially enriched between the patient groups. For the annotation of protein pathways, we used the protein subsystem classification of the SEED genome annotation database (2). The annotation revealed 971 protein subsystems of which 319 had a minimum abundance of 0.1% across all samples which we used for detailed analysis. We identified 10 protein annotation pathways to be statistically different between the patient groups. An exciting part of the analysis was to identify any correlation between protein annotation pathways and metagenomic shotgun sequenced species. Remarkably, several protein annotation pathways were found to be significantly enriched in GPA patients compared to the healthy control group, amongst correlation between other groups. The two most interesting and discussed pathway in our main paper were an enriched pathway of chorismite synthesis in GPA and disease control patients and an association with enrichment of genes involved in the synthesis of vitamin B12 in GPA and their house hold controls compared to the disease controls and healthy controls. Why there is an enrichment of genes involved in Vitamin B12 biosynthesis in GPA patients is not clear but we proposed a hypothesis in our main paper.
In summary, our combinational approach of bacterial 16S gene sequencing and deep shotgun sequencing of the nasal microbiome from GPA patients and controls, revealed several exciting insights with a unique Staphylococcus species profile and protein pathway associated into the complex disease. Our study also highlights the power of using shotgun sequencing for a more complex investigation of the human microbiome with bacterial protein subsystem.
- Yarbrough ML, Lainhart W, Burnham CA: Epidemiology, Clinical Characteristics, and Antimicrobial Susceptibility Profiles of Human Clinical Isolates of Staphylococcus intermedius Group. J Clin Microbiol 2018, 56(3).
- Overbeek R, Olson R, Pusch GD, et al. The SEED and the Rapid Annotation of microbial genomes using Subsystems Technology (RAST). Nucleic Acids Res. 2014 Jan;42(Database issue):D206-14. doi: 10.1093/nar/gkt1226.
Follow the Topic
-
Microbiome
This journal hopes to integrate researchers with common scientific objectives across a broad cross-section of sub-disciplines within microbial ecology. It covers studies of microbiomes colonizing humans, animals, plants or the environment, both built and natural or manipulated, as in agriculture.

Related Collections
With Collections, you can get published faster and increase your visibility.
Oncobiome
This collection of papers delves into the burgeoning field of oncobiome research, exploring the intricate relationship between cancer and the microbiome. The oncobiome encompasses the diverse microbial communities residing in and on the human body, which influence cancer development, progression, and treatment responses. By examining these interactions, our aim is to unravel the complex mechanisms through which the microbiome impacts oncogenesis and therapeutic outcomes.
This compilation highlights cutting-edge research, offering insights into potential diagnostic markers and novel therapeutic strategies, thereby advancing our understanding of cancer biology and paving the way for innovative, microbiome-targeted cancer treatments.
This is a cross-journal collection between:
Experimental Hematology and Oncology
Articles will undergo the standard peer-review process of the journal to which they are submitted and are subject to either the BMC editorial policies or those of BJC Reports. Articles will be added to the Collection as they are published. The Editors have no competing interests with the submissions which they handle through the peer review process. The peer review of any submissions for which the Editors have competing interests is handled by another Editorial Board Member who has no competing interests.
Publishing Model: Open Access
Deadline: Ongoing
Animal Gut Nutrition and Greenhouse Gas Mitigation
Animal Microbiome, Journal of Animal Science and Biotechnology and Microbiome call for submissions to the collection on Animal Gut Nutrition and Greenhouse Gas Mitigation.
Efforts to reduce greenhouse gas emissions from livestock systems increasingly hinge on innovations in animal gut nutrition. The dynamic relationship between the gut microbiome and nutrient utilization plays a pivotal role in shaping methane output, feed efficiency, and overall sustainability. Advances in microbial ecology—particularly in understanding the role of gut microbiome in nutrient metabolism—are opening new pathways for mitigating emissions while enhancing productivity. These developments support the implementation of climate-smart agricultural strategies to address climate change and its impacts.
Looking ahead, continued research in this field has the potential to yield innovative solutions such as targeted probiotic supplementation, which could further optimize gut function and enhance nutrient absorption. These advancements may lead to reduced greenhouse gas emissions while improving animal health and productivity. By deepening our understanding of the animal gut microbiome, we can contribute significantly to sustainable agricultural practices that benefit both the environment and food security.
We invite researchers to contribute to this special Collection on Animal Gut Nutrition and Greenhouse Gas Mitigation. Topics of interest include but are not limited to:
- Animal Gut Microbiome and Feed Efficiency
- Greenhouse Gas Mitigation Strategies
- Rumen Fermentation Dynamics
- Nutrient Utilization in Livestock
- Probiotic Supplementation Effects
- Sustainable Livestock Production Practices
- Climate-Smart Agriculture Innovations
This Collection supports and amplifies research related to SDG 13, Climate action.
All submissions in this collection undergo the relevant journal’s standard peer review process. Similarly, all manuscripts authored by a Guest Editor(s) will be handled by the Editor-in-Chief of the relevant journal. As an open access publication, participating journals levy an article processing fee (Animal Microbiome fees, Journal of Animal Science and Biotechnology fees, Microbiome fees). We recognize that many key stakeholders may not have access to such resources and are committed to supporting participation in this issue wherever resources are a barrier. For more information about what support may be available, please visit OA funding and support, or email OAfundingpolicy@springernature.com or the Editor-in-Chief of the journal where the article is being submitted.
Publishing Model: Open Access
Deadline: Sep 04, 2026


Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in