When Silence Speaks: How BRCA1 Hypermethylation Fuels Sporadic Breast Cancers
Published in Cancer
Decoding BRCA1 promoter hypermethylation: a new frontier in understanding sporadic breast cancer
DOI: 10.1038/s41417-025-00969-7.
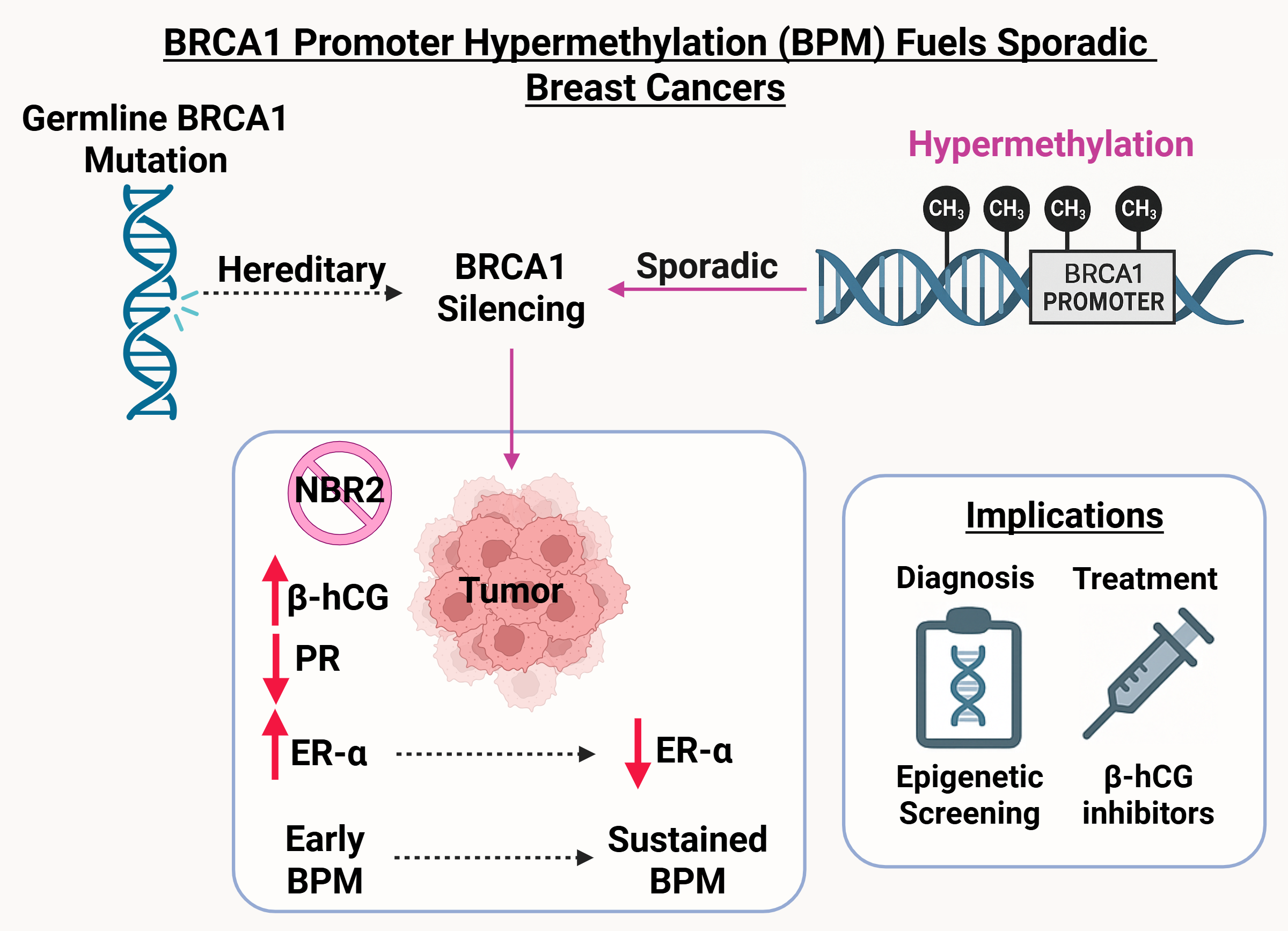
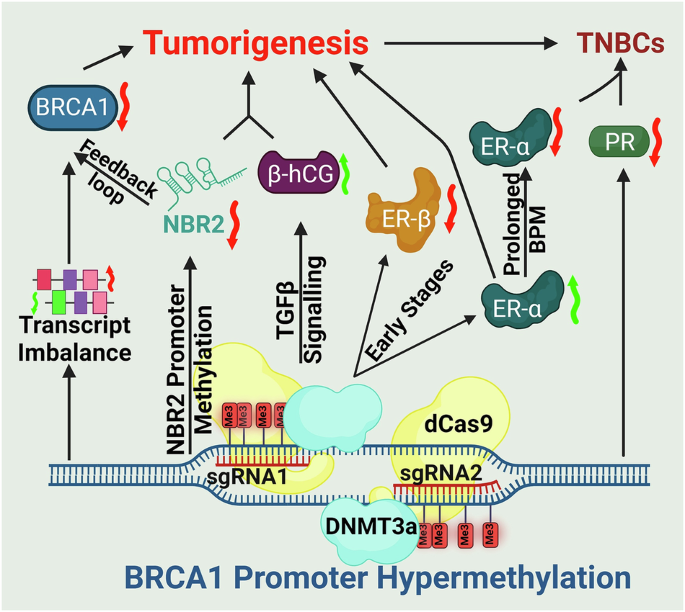
Germline BRCA1 mutations are a defining feature of hereditary breast and ovarian cancers, yet they account for only a fraction of overall breast cancer cases. In contrast, most patients present with sporadic tumors, where the molecular basis of BRCA1 deficiency has remained less clear. Intriguingly, many of these cancers exhibit biological and clinical features that mirror BRCA1-mutated cases. This suggests that epigenetic silencing could potentially be an alternative mechanism of gene inactivation. Our study establishes BRCA1 promoter hypermethylation (BPM) as one such mechanism, acting as a functional parallel to inherited mutation.
Although BPM has been described in patient samples, its causal role in tumor development had not been systematically demonstrated. We addressed this challenge by creating a targeted system to induce BRCA1 promoter hypermethylation in human breast cancer cells. This approach allowed us to disentangle correlation from causation and directly observe how epigenetic silencing reshapes transcriptional programs, hormone receptor signaling, and tumor behavior.
A multifaceted impact on tumor biology
Our analysis uncovered three interlocking features of BPM-driven oncogenesis:
- Coupled repression of NBR2: Because BRCA1 and the lncRNA NBR2 share a promoter, hypermethylation silences both. The resulting loss of NBR2 disrupts AMPK signaling and establishes a feedback loop that amplifies BRCA1 downregulation.
- Hormone receptor remodeling: BPM initiates a temporal switch, initially augmenting ER-α–mediated DNA repair and proliferation, but ultimately driving tumors toward a hormone receptor–negative phenotype characteristic of triple-negative breast cancers.
- Immune microenvironment modulation: BPM induces β-hCG, a factor implicated in immune suppression and resistance pathways, thereby linking epigenetic silencing to tumor–immune crosstalk.
Translational significance
In xenograft models, BPM tumors displayed enhanced growth, poor differentiation, and invasive histological features. Proteomic analyses identified stage-specific molecular signatures and candidate biomarkers, including HSP90, STAT1, SPEN, TFF1, and β-hCG. These results highlight the potential of BPM to serve as:
- a biomarker for patient stratification, complementing BRCA1 mutation testing,
- a prognostic indicator of tumor aggressiveness, and
- a therapeutic entry point, whether through epigenetic reactivation strategies or targeting downstream effectors such as β-hCG or HSP90.
Expanding the paradigm of cancer epigenetics
By establishing promoter hypermethylation as a recurrent and functionally significant mode of BRCA1 loss, this work underscores a broader principle: epigenetic lesions can phenocopy genetic mutations and drive tumorigenesis through convergent pathways. This recognition has practical implications as it expands the population of patients who may benefit from DNA damage–targeted therapies and underscores the need to integrate epigenetic profiling into precision oncology frameworks.
Our findings reinforce the importance of treating epigenetic events not as ancillary changes, but as core drivers of tumor evolution. Recognizing and targeting such epigenetic lesions is essential for comprehensive cancer diagnosis and treatment. We anticipate that future efforts will build upon these insights to develop methylation-based biomarkers and explore therapeutic strategies that restore BRCA1 function or exploit vulnerabilities created by its epigenetic silencing. In doing so, the field moves closer to extending BRCA1-targeted strategies to a far broader patient population than previously possible.
Follow the Topic
-
Cancer Gene Therapy
The essential gene and cellular therapy resource for cancer researchers and clinicians, keeping readers up to date with the latest developments in gene and cellular therapies for cancer.



Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in