

Infections caused by fungi pose a serious health threat, affecting 1,000 million people and causing 1.5 million deaths each year worldwide. The problem has been growing over the last decades as more people received medical treatments that predispose towards such infections. For instance, patients undergoing chemotherapy or immunosuppressive treatments after organ transplants have impaired immune systems, making them particularly vulnerable. From a clinical perspective, treating fungal infections is difficult because available antifungal drugs are expensive and have undesired side effects. In addition, the incidence of drug-resistant strains is on the rise, which often leads to therapeutic failure. Thus, there is a need to improve current treatments for fungal infections. A promising strategy to achieve this is to study how these pathogens have adapted, from an evolutionary (genomic) perspective, to the human host and to antifungal drugs. This may help us understand the molecular mechanisms of virulence and drug resistance, leading to better therapies in the future. In this work we addressed this issue for Candida pathogens, a group of yeasts that are the primary cause of hospital-acquired fungal infections.
For many years, our group has been interested in how virulence traits have emerged multiple independent times across Candida and other pathogens, mostly by comparing the genomes of different fungal species. We figured out that the growing amount of genome sequences deposited in public databases provided a unique opportunity to ‘zoom in’ and study the most recent steps of this evolutionary process, happening between strains of a given species. We analysed ~2,000 public genomes from clinical isolates of six major Candida pathogens sampled around the world (C. glabrata, C. auris, C. albicans, C. tropicalis, C. parapsilosis and C. orthopsilosis). These genomes were compared to a reference to obtain a catalogue of genetic variants, which enabled the study of recent adaptation within each of these six species. Notably, we analysed the role of both simple variants (i.e. Single Nucleotide Polymorphisms) and complex structural variants (i.e. inversions or translocations), which have been mostly overlooked in Candida.
On the one hand, we explored the genes with signs of recent selection in our genomic datasets, which inform about biological processes underlying adaptation to human-related environments. Such selection measurements are typically done by finding genes with an excess of genetic variants, but this is not so straightforward for Candida pathogens because they have unique ecological niches. Most of them are opportunistic, meaning that they thrive as harmless human commensals or in other environments, such as the gut and skin of wild animals, the bark of forest trees or warm sea waters. Only in particular contexts - e.g. when colonising individuals with impaired immune systems - they adopt a pathogenic behaviour. Thus, many genetic variants present in clinical isolates may reflect environmental selective pressures, unrelated to human adaptation. To truly understand human-related adaptation it was necessary to analyse only variants appearing in a clinical context. We reasoned that such variants should be among the most recently-acquired ones by clinical isolates, as they appeared closer to the context of isolation. To approximate that, we considered variants that appeared within clusters of clinical isolates being genetically very closely related. We found hundreds of genes with an excess of such recent variants, which we interpret as genes affected by recent, clinically-relevant selection. These involve highly diverse biological processes which are mostly unique to each Candida species. In addition, we find that cell adhesion has been shaped by selection in multiple species, indicating that it is a very important factor for human-related adaptation.
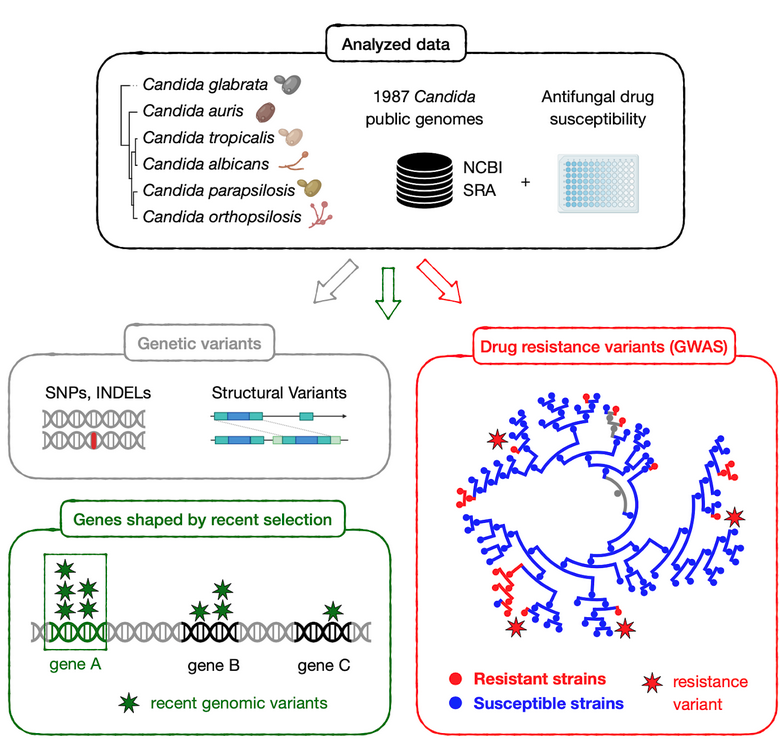
On the other hand, we used our dataset to find genetic variants underlying drug resistance. In recent years our group has been studying this phenomenon by evolving drug-resistant strains in the lab, to then identify genetic variants that appear during this artificial evolution. This has provided important insights into the evolutionary, genetic mechanisms of drug resistance across various Candida species. However, these approaches rely on in vitro models which may not entirely reflect the process of drug adaptation in the clinics. To solve this, and identify the variants driving resistance in clinical isolates, we combined our genomic data with information about drug susceptibility for the sequenced isolates, and performed a Genome-Wide Association Study (GWAS). Essentially, we systematically screened for variants associated with antifungal drug resistance, which revealed both known and new mechanisms of resistance towards seven antifungal drugs in three Candida species. Furthermore, our analysis suggests an unexpected, but worrying, finding: resistance may sometimes spread due to sex between susceptible and resistant strains, which may partially explain why it is so prevalent among Candida pathogens.
In summary, our work illuminates how these pathogens adapted to humans and to antifungal drugs. Beyond the new knowledge generated, our catalogue of variants, selection signatures and drivers of drug resistance is a valuable resource for future confirmatory experiments, which could lead to better treatments for Candida infections.
Follow the Topic
-
Nature Microbiology

An online-only monthly journal interested in all aspects of microorganisms, be it their evolution, physiology and cell biology; their interactions with each other, with a host or with an environment; or their societal significance.
Related Collections
With Collections, you can get published faster and increase your visibility.
The Clinical Microbiome
Publishing Model: Hybrid
Deadline: Mar 11, 2026


Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in