16S rRNA and Shotgun Together At Last with Greengenes2
Published in Microbiology
Explore the Research

Greengenes2 unifies microbial data in a single reference tree - Nature Biotechnology
A comprehensive microbial resource reconciles genomic and 16S rRNA data in a single tree.
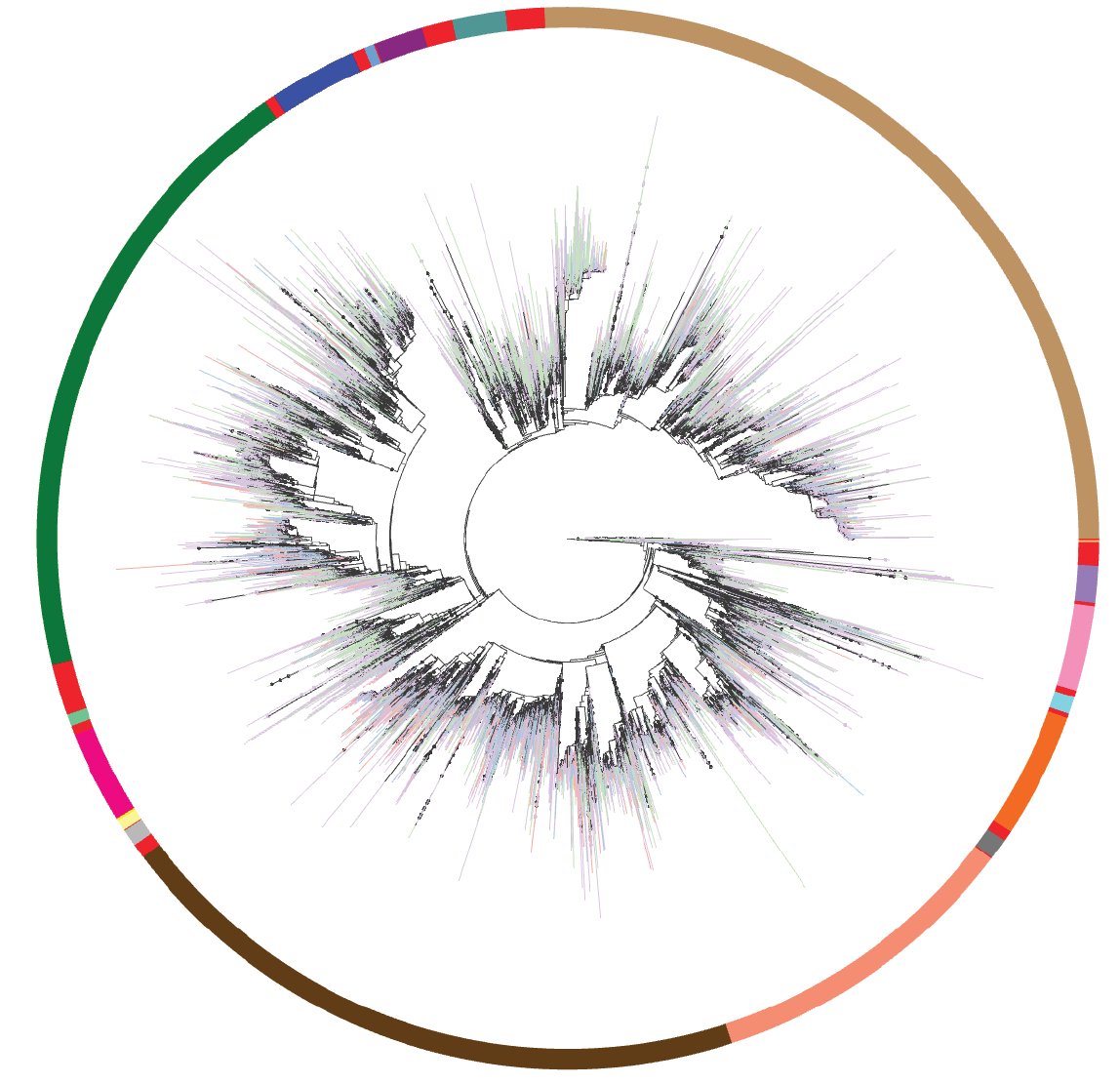
The Greengenes reference database has a long history of use for characterizing microbial communities (see McDonald et al. ISMEJ 2011 and DeSantis et al. AEM 2006). It was the 16S rRNA reference used for massive endeavors such as the Human Microbiome Project, the American Gut Project and the Earth Microbiome Project, and numerous other studies over the past many years. One of the critical roles that Greengenes occupies is providing a standard taxonomy, and phylogeny, which can be used to interpret what organisms are present in a microbiome sample. The Greengenes taxonomy is coupled to the phylogeny, allowing for recovery of unlabeled environmental data, and the expression of taxon labels reflective of an unsupervised objective evolutionary hypothesis of the sequence relationships. Within microbiome analysis, the phylogeny further provides support for common diversity calculations such as Faith's Phylogenetic Diversity and UniFrac, as well as predictive techniques like PICRUSt.
Here we provide the first major update to Greengenes in a decade, called Greengenes2, where we substantially revise the construction of the database and usher in new capabilities for the analysis of microbiome data. Specifically, we integrated high quality full length 16S rRNA sequences into a whole genome phylogeny using a novel algorithm called uDance. We then placed over 20,000,000 16S rRNA V4 amplicon sequence variants, and mitochondria and chloroplast from SILVA, into the phylogeny using a different novel algorithm called DEPP. These steps yield a single phylogeny that expresses both genomes and 16S rRNA data. We then decorated a taxonomy based on the GTDB and Living Tree Project onto the phylogeny using an adaption of the original tax2tree algorithm to characterize the database.
This approach allows us to combine 16S rRNA and shotgun sequencing samples together with substantially reduced technical bias. It was traditionally assumed these different molecular preparations would inhibit combined analysis and that these techniques were prone to tell different stories. However, we demonstrate that these biases can be overcome through application of computational techniques against a common database.
Our first hint that these biases could be overcome came from a principal coordinates analyses on human fecal specimens sequenced for both 16S rRNA V4 amplicons and shotgun metagenomics (figure 1). In the absence of phylogeny, we observe a strong technical separation (this is true even if collapsed to genus level), however we are able to remove the technical separation by leveraging the phylogeny with UniFrac.
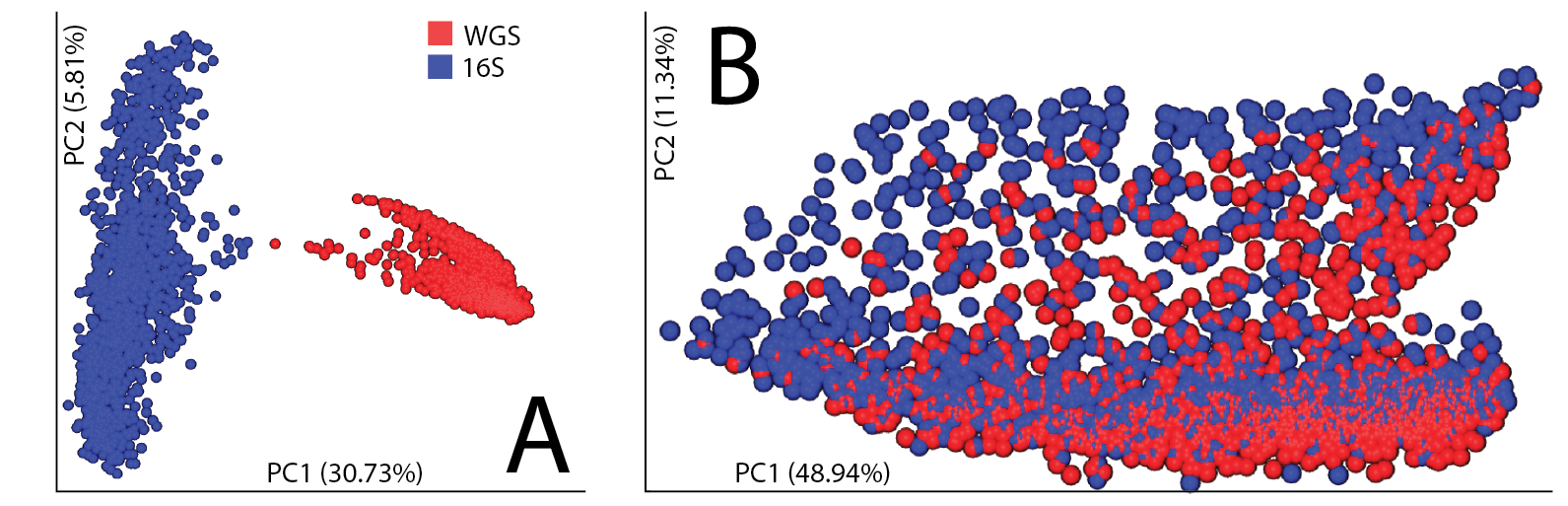
Figure 1, integrating human fecal samples sequenced for both 16S and shotgun. (A) Beta diversity assessed using Bray-Curtis followed by principal coordinates analysis. (B) Beta diversity assessed using weighted UniFrac followed by principal coordinates analysis.
We then tested for consistency in taxonomic profiles. In doing so, we considered two approaches to obtain taxonomy for 16S V4 amplicons. First, we trained a traditional Naive Bayes classifier on the full length 16S. Second, we utilized the taxonomy directly from the phylogeny based on the placements of the individual amplicon sequence variants (ASVs). Excitingly, we not only observed a high level of correlation between the 16S and shotgun data, but that the phylogenetic approach outperformed the Naive Bayes classification! And, remarkably, we obtain reasonable correlation even at species level. While care must be taken in providing species level information with 16S (even full length), it is noteworthy that it appears some evidence for species assignment relative to this database may be attainable (figure 2).
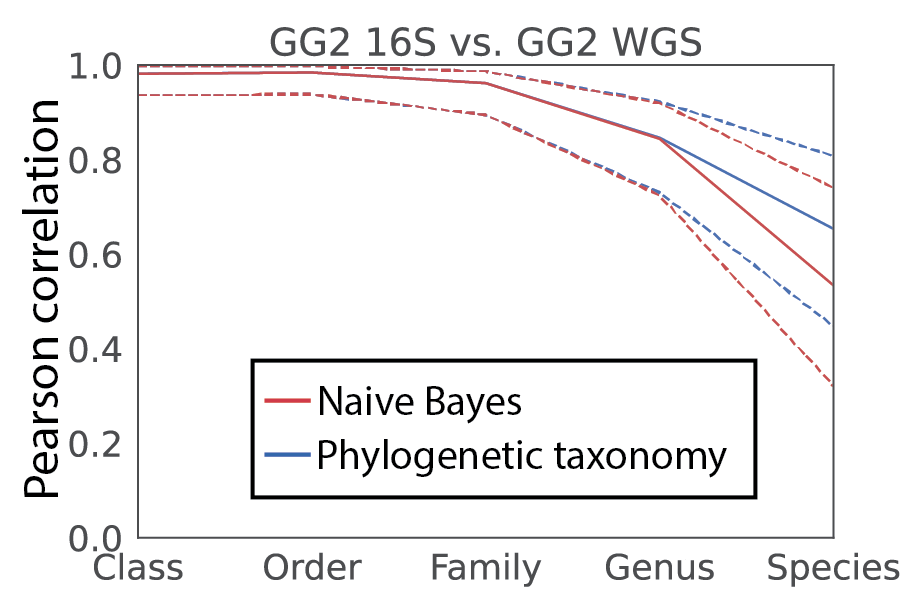
Figure 2, Pearson correlation of taxonomy profiles for human fecal samples sequenced for both 16S and metagenomics. The Naive Bayes classifier used was trained on the full length 16S data (genus r=0.84; species r=0.54). The phylogenetic taxonomy utilized amplicon sequence variant fragment placements (genus r=0.85; species r=0.65).
Perhaps the most important result though was the observed consistency in effect size calculations. Using the paired 16S and shotgun samples again, we computed effect sizes off of beta diversity using Evident across a large number of study covariates for both data layers. We specifically considered Bray-Curtis and weighted UniFrac (figure 3). We used Bray-Curtis, as to the best of our knowledge, non-phylogenetic measures are the "state-of-the-art" for beta-diversity comparisons of 16S and shotgun. Amazingly, weighted UniFrac resulted in an r2 of 0.86, suggesting that the 16S and shotgun data are largely telling the same story!
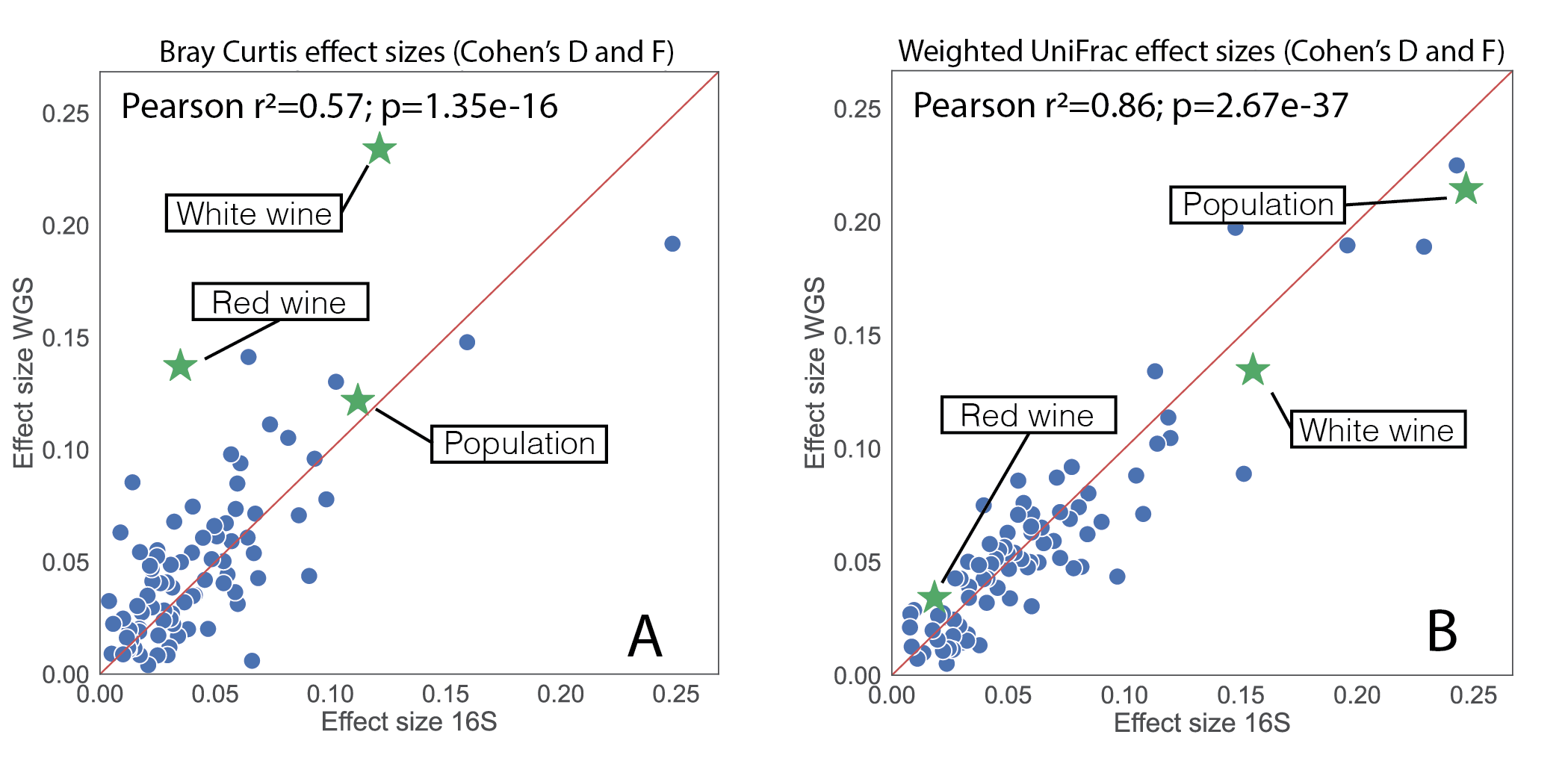
Figure 3, effect size calculations on paired 16S and metagenomic samples. (A) effect sizes when Bray Curtis was used to assess beta diversity. (B) effect sizes using weighted UniFrac which accounts for phylogeny in beta diversity assessment.
These data suggest that it is feasible to rescue the millions of microbiome samples sequenced for 16S rRNA amplicons, and that the field can continue to leverage those historical data in context to modern shotgun metagenomic techniques. This apparent consistency between 16S and shotgun enables tests of reproducibility with these different molecular methods.
While the primary results discussed are on human fecal samples, which are more densely sampled for whole genomes than other environments, we also demonstrate reduced technical bias in the recent Earth Microbiome Project dataset with its paired 16S and shotgun samples -- this is noted in the supplemental of the Greengenes2 Nature Biotechnology publication.
The recent release of Greengenes2 is an important milestone, not an endpoint. There is a vast amount of genomic information not yet represented by any database, let alone Greengenes2, and it is critical that we focus on continued growth. Separately, taxonomy is a dynamic entity, therefore it is imperative mechanisms exist to simplify updates. We are keenly aware of these concerns, and are hard at work to ensure long term growth and stability of this resource. We have further exciting updates coming, and we are eager to collaborate on use, growth, and development. For inquiries, please do not hesitate to contact me at d3mcdonald@eng.ucsd.edu.
Greengenes2 is a BSD-licensed reference database. This is a permissive license suitable for academic and industry alike. Flat and QIIME 2 compatible files can be found on our official FTP. A tutorial on using Greengenes2 with QIIME 2 can be found on the QIIME 2 Forum. We host an interactive, but limited, website for exploring Greengenes2 which of note provides observed environmental information for 16S V4 ASVs based on existing sample metadata. The primary scripts that construct the database are on GitHub. And finally, you can read about the resource in our Nature Biotechnology publication.
This post was generously copyedited by Dr. Celeste Allaband, DVM, PhD.
My background is computational, and I emphasize open source software development and open access data. I lead Greengenes, BIOM-Format, redbiom, and in recent years UniFrac. I am the Scientific Director for the American Gut Project / Microsetta Initiative, and am a major contributor to projects like QIIME, Qiita and the Earth Microbiome Project. Within QIIME 2, I am a regular contributor on the forum and can be reached easily there under user @wasade. On GitHub, I can also be found under @wasade.
Follow the Topic
-
Nature Biotechnology
A monthly journal covering the science and business of biotechnology, with new concepts in technology/methodology of relevance to the biological, biomedical, agricultural and environmental sciences.




Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in