A crystal graph neural network model for defect formation in clean energy materials
Published in Computational Sciences
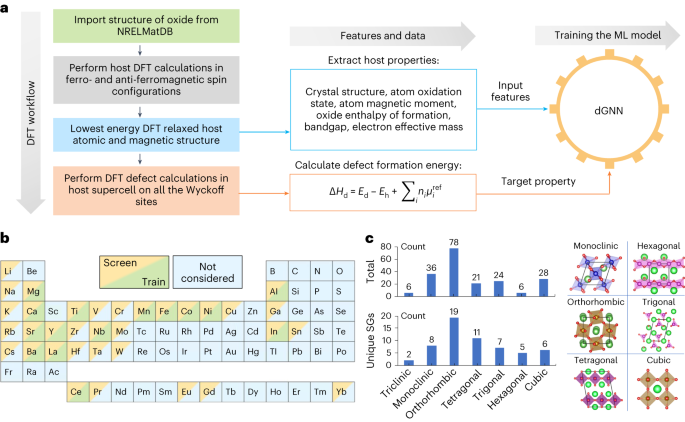
The properties and functionality of solid-state materials are governed by their chemical composition and crystal structure. Often, however, it is not only the ideal crystal structure that matters but also the material’s ability to form defects. This is especially true in oxides for renewable energy application, where defects enable, for example, the ionic conductivity in solid electrolytes or the redox processes for solar thermochemical hydrogen (STCH) generation.
To efficiently screen a large number of materials for such applications, we were seeking after a machine learning (ML) model that would predict the propensity of materials to form these enabling defects, avoiding the need for a very large number (possibly up to a million) of comparatively slow and tedious density functional theory (DFT) supercell calculations. Graph neural networks (GNNs) are promising for this task because they automatically extract crystal structure information as features for model inputs, and therefore go beyond the capabilities of simpler models based only on chemical and compositional characteristics that can be derived by hand. However, existing GNN models were primarily designed to describe the ideal crystal without defects. Therefore, they predict the properties of the entire input structure, not site-specific properties as desired here.
The basic premise of our work is that the properties of defects, such as O vacancies, should be encoded in the crystal structure of the ideal oxide. Thus, an ML model could predict the vacancy formation energies without the need to determine the supercell structure of the defected material beforehand. We implemented node-level pooling operations for our defect-GNN model (dGNN) which extracts site-specific properties on an underlying crystal structure. To generate the training data for the ML model, we performed DFT supercell calculations to obtain defect formation energies for ~1500 unique defect sites in about 200 different oxides representing a chemical space of 15 different metal cations. Extensive cross-validation statistics ensured the numerical robustness and demonstrated that the model is capable of digesting future additional training data to further increase the accuracy.

The dGNN model enables us to screen large databases. As a first exercise, we applied the model on over 2000 oxides in the Materials Project database that match the elements of the training set. Progressively stringent multi-objective selection criteria narrow this space down to a manageable number of candidates that are worthwhile for further investigation. We also connected the database screening with thermodynamic modeling, facilitating the creation of experimentally relevant temperature-pressure diagrams and analyzing the reduction entropy as an additional selection criterion in a high-throughput fashion.
The dGNN approach is a versatile framework for all crystal/symmetry classes and chemistries without need for engineering of descriptors and feature vectors. It is generally independent of the underlying graph encoding and convolutional architectures, so that it can be readily integrated with new developments in these areas. The defect-data and the codes implementing the dGNN approach are publicly available in Zenodo and Github repositories. We envision a broadening of the current chemical space, providing access to much larger number of candidate materials, as well as future extensions to more complex defects that could further enhance the materials performance in clean energy technologies.
Original research paper: Matthew D. Witman, Anuj Goyal, Tadashi Ogitsu, Anthony H. McDaniel, and Stephan Lany. Defect graph neural networks for materials discovery in high-temperature clean-energy applications. Nat. Comput. Sci. (2023).
Follow the Topic
-
Nature Computational Science
A multidisciplinary journal that focuses on the development and use of computational techniques and mathematical models, as well as their application to address complex problems across a range of scientific disciplines.



Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in