A designer peptide against the EAG2–Kvβ2 potassium channel targets the interaction of cancer cells and neurons to treat glioblastoma
Published in Cancer
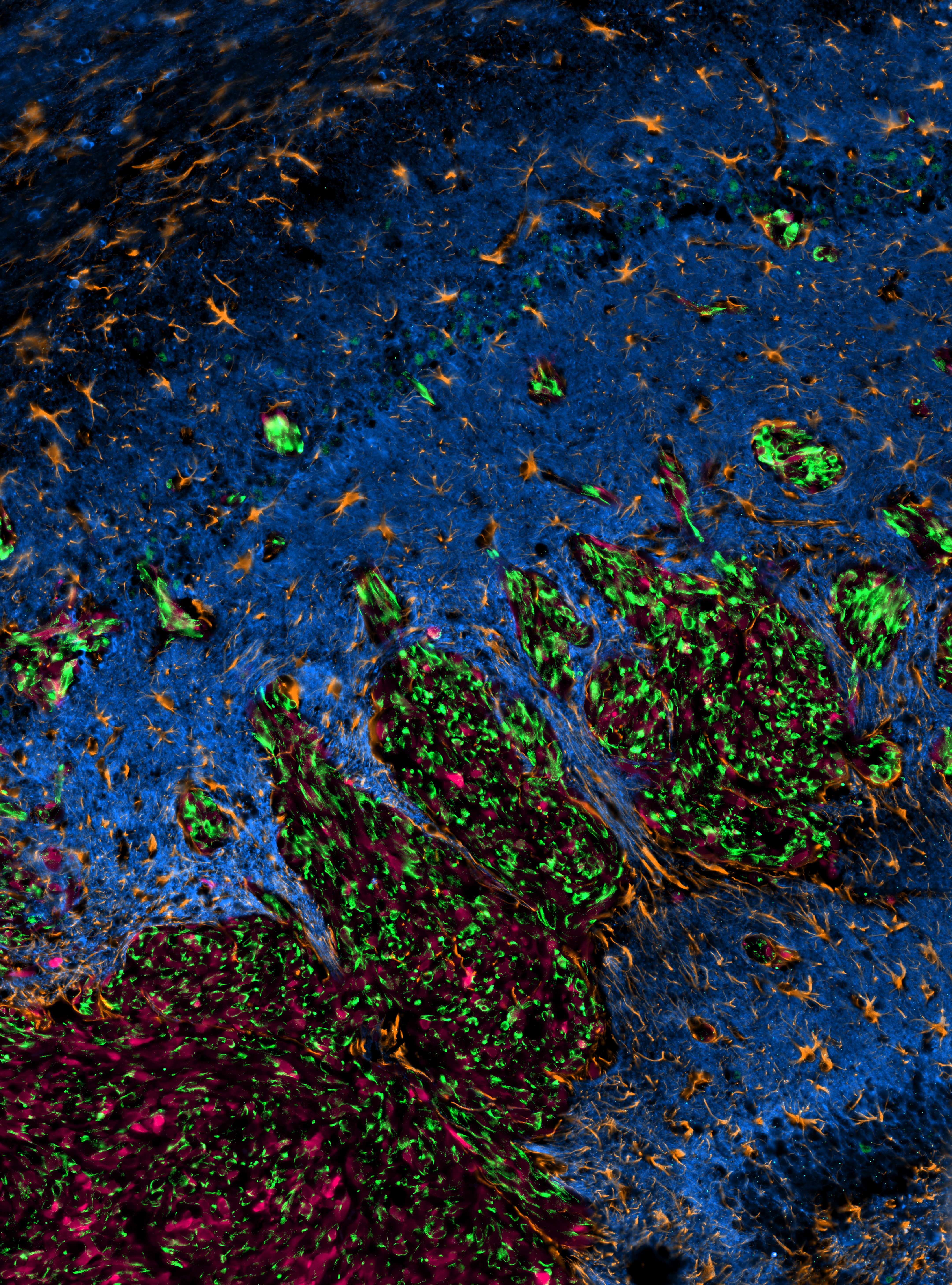

Glioblastoma (GBM) is the most common and aggressive brain cancer1. Recent groundbreaking findings have illuminated the interactions between GBM cells and neurons. Both paracrine communications and direct synaptic connections can occur between neurons and GBM cells. These connections allow neuronal inputs to initiate calcium transients in GBM cells, facilitating tumor growth, invasion, and therapy resistance2,3. While previous studies have unearthed the intricacies of neuron-GBM interactions, therapeutic approaches that target this tumor-promoting mechanism remain elusive.
Temozolomide (TMZ) is the standard-of-care chemotherapy to treat GBM. Though it offers patients an extended survival of ~2.5 months4, this (very limited) benefit comes with a price. The toxic effects of TMZ are evident in the neural, gastrointestinal, and hematopoietic systems, stemming from TMZ's inability to discern tumor cells from healthy cells. Additionally, over half of GBM patients either present upfront TMZ resistance or develop it over time5. These challenges underscore the pressing need to identify vulnerabilities that are uniquely present in cancer cells and develop rational-based therapy to address these GBM weaknesses.
In a recent study, we discovered a tumor-promoting potassium channel complex that appears to only form in GBM cells and invented a designer peptide that can not only disrupt this channel complex, but also effectively treat TMZ-resistant GBM, opening a fundamentally new avenue for GBM treatment.
EAG2-Kvβ2 potassium channel complex regulates neuron-GBM interaction
Delving into the Ivy GBM Atlas Project dataset (a spatially revolved RNA sequencing database)6, we focused on the tumor-brain interface—the prime location for neuron-GBM interaction. A voltage-gated potassium channel, EAG2, and a potassium channel-associated protein, Kvβ2, stood out for their high expression in GBM cells at this region. This observation was corroborated by immunostaining using multiple patient-derived xenograft GBM models. When we knocked down these two proteins, we observed a marked reduction in tumor volume, a pronounced decline in tumor invasiveness, and significantly increased mouse survival. These compelling data set the stage for our deeper dive into the roles of EAG2 and Kvβ2. First, we employed a neuron-GBM cell coculture system. When GBM cells physically contact the neurites extended from neurons, EAG2 predominantly localizes at cell-cell contact regions, whereas EAG2 displays widespread distribution in GBM cells that are cultured alone or not in contact with neurons. This observation suggests that EAG2 is actively recruited to the cell-cell contact site. Indeed, the neuronal contact-dependent localization of EAG2 was hampered by Kvβ2 knockdown in GBM cells. EAG2 knockdown decreases calcium transients in GBM cells, uncovering a role of EAG2 in governing neuron-driven calcium signaling in GBM cells. Indeed, knockdown of EAG2 or Kvβ2 hindered neuron-driven GBM cell growth, further highlighting their pivotal roles in neuron-GBM interaction.
EAG2 and Kvβ2 form a unique interaction with therapeutic potential
We next asked: do EAG2 and Kvβ2 form a functional potassium channel complex? Consistent with our observation that EAG2 knockdown and Kvβ2 knockdown phenocopy each other, we detected physical interaction between EAG2 and Kvβ2 in a panel of patient derived GBM cell lines that represent the heterogeneous GBM subtypes. Quite surprisingly, their interaction was undetectable in human fetal neural stem cells or protein lysate from the entire normal adult mouse brains. Such a distinction suggests immense translational potential. We next set to uncover the cause behind this GBM-specific protein-protein interaction (PPI). After exploring a myriad of possibilities, we made a breakthrough: the 4th isoform of Kvβ2 protein (Kvβ2iso4), but not Kvβ2 isoforms 1, 2, 3, or 5, is able to physically interact with EAG2. When we co-expressed EAG2 with Kvβ2iso4, we observed a marked increase in the overall cellular potassium channel activity. Adding to the intrigue, Kvβ2iso4 is highly expressed in human GBM cells but not a list of other human cell types that we tested, including human fetal neural stem cells, human astrocytes, HEK293T cells, and two lung cancer cell lines.
Together, we identified a GBM-specific PPI that is required for neuron-driven GBM growth. The specificity of this interaction suggests the exciting possibility that targeting EAG2-Kvβ2 PPI may mitigate GBM growth with minimal side effects (because this PPI seems to be absent in non-cancer cells).
A designer peptide with remarkable promise to treat GBM
To develop a rational-based therapy, we decided to design peptides, which are comprised of various lengths of amino acid sequences that match the Kvβ2 amino acid sequence required for its interaction with EAG2. We postulated that these designer peptides can competitively interfere with EAG2-Kvβ2 interaction. After painstakingly truncating the Kvβ2 protein and biochemically characterizing each truncation mutant, we identified a critical Kvβ2 domain to inform our peptide design. One designer peptide, which matches the amino acid residues 90 to 114 of Kvβ2, showed exceptional efficacy in blocking EAG2-Kvβ2 PPI and neuron-driven GBM cell growth in the coculture system. Encouraged by these results, we moved on to test the designer peptide’s therapeutic efficacy in mice.
By using an osmotic pump for direct intratumoral peptide delivery, we obtained remarkable results: the designer peptide treatment pronouncedly reduced tumor size, decreased tumor invasion, and extended mouse survival in a panel of patient-derived xenograft models and a syngeneic allograft model of GBM that represent all major GBM subtypes.
Moreover, our single cell analysis identified the specific population of GBM cells targeted by the designer peptide. These tumor cells display gene expression signatures that not only support their interactions with neurons but also indicate that they harbor TMZ resistance. This raised a tantalizing possibility: Could our peptide target the notorious TMZ-resistant GBM cells? Indeed, our designer peptide is highly efficacious in mitigating the growth of TMZ-resistant GBM in two xenograft models!
Our journey has not only furthered our understanding of neuron-GBM interactions but also introduced a potential game-changer for GBM treatment (Figure 1). By actively advancing our designer peptide towards clinical trials, we hope to develop a new therapy to benefit GBM patients in the not-too-distant future.
.png)
Figure 1. EAG2 and Kvβ2 physically interact to form a potassium channel complex at neuron-GBM contact sites. EAG2-Kvβ2 complex regulates Ca2+ transients of GBM cells and promotes GBM growth, invasion, and chemoresistance. Designer peptide K90-114TAT disrupts EAG2-Kvβ2 complex formation, mitigates GBM aggression, and is efficacious in treating both TMZ-sensitive and -resistant GBM.
Reference
- Kotecha, R., Odia, Y., Khosla, A. A. & Ahluwalia, M. S. Key Clinical Principles in the Management of Glioblastoma. JCO Oncol Pract 19, 180–189 (2023).
- Mancusi, R. & Monje, M. The neuroscience of cancer. Nature 618, 467–479 (2023).
- Winkler, F. et al. Cancer neuroscience: State of the field, emerging directions. Cell 186, 1689–1707 (2023).
- Stupp, R. et al. Radiotherapy plus Concomitant and Adjuvant Temozolomide for Glioblastoma. www.nejm.org.
- Lee, S. Y. Temozolomide resistance in glioblastoma multiforme. Genes and Diseases vol. 3 198–210 Preprint at https://doi.org/10.1016/j.gendis.2016.04.007 (2016).
- Puchalski, R. B. et al. An anatomic transcriptional atlas of human glioblastoma. Science (1979) 360, 660–663 (2018).
Follow the Topic
-
Nature Cancer
This journal aims to provide a unique forum through which the cancer community will learn about the latest, most significant cancer-related advances across the life, physical, applied and social sciences.

Related Collections
With Collections, you can get published faster and increase your visibility.
Cancer Neuroscience: from mechanisms to therapy
Publishing Model: Hybrid
Deadline: Jan 30, 2027


Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in