A genotype imputation reference panel specific for native Southeast Asian populations
Published in Genetics & Genomics
Capturing as many variations as possible in a population has been one of the main tenets of population genetics. With a large enough number of variants in a dataset, information about a population can be inferred more precisely. However, capturing these variants has also been challenging. Typically, genotyping arrays are applied in most genonic studies. However, they are primarily based on major populations of the world, particularly the Europeans. This often results in the marginalized populations from other parts of the world being underrepresented, thus losing the putative genetic variations that could be captured. Losing these markers further undermines the power to detect natural selection signals, or association signals for disease studies.
To address this issue, imputation is commonly used. It is a statistical inference of ungenotyped SNPs using a much larger dataset as a reference panel, typically comprising sequencing data of hundreds to thousands of samples. This method works by matching haplotypes found in a study dataset with haplotypes available in a reference panel and assigning probabilistic genotypes that previously were not observed. The mechanism of imputation favors a condition where a similar stretch of haplotype is found between the reference panel and target dataset, allowing a SNP to be predicted. Thus, imputation is presumably reference panel sensitive. However, there is currently a lack of evidence-based evaluation on how far the choice of reference panel impacts the performance of imputation, albeit a larger reference panel translates into better imputation performance. In the case of currently available mainstream reference panels, the population composition is saturated with the European, and to a lesser extent, the East Asian ancestry, while other populations are severely underrepresented. For instance, while Southeast Asia (SEA) populations are among the most diverse populations, and represent almost 10% of the global population, their representation in the major publicly available panels is neglected. Although, currently there are consortium studies such as GA100K Pilot which encompasses the majority of the Asian population, its effectiveness and accuracy to be used as a reference panel for a wider range of SEA populations has yet to be determined. The limited choice of reference panel may potentially limit the accuracy and power when imputing populations that are not represented, especially in the imputation of rare variants, where frequency between populations could vary significantly. Rare variants and population-specific variants are of significance in disease-based studies because they often carry higher effect sizes and may advance our understanding of disease predisposition within a population. Furthermore, with the availability of numerous options for imputation tool pipelines, each with its strengths and constraints, yet it can be a dilemma when deciding on an ideal option.
Orang Asli (OA) is of particular interest in population genetics in SEA due to its long population history and unique genotypic and phenotypic characteristics. They are postulated to be among the earliest anatomically modern humans that populated SEA. Throughout their long history of isolation, the OA populations are expected to harbor unique genetic variants that not only allow them to locally adapt to the harsh environment of tropical rainforests, but also contribute to their distinct morphology and appearance as opposed to the mainstream populations in the region. However, owing to various constraints, large-scale population genomic and sequencing projects for OA have been underrepresented.
In this study, we evaluated the performance of imputation using a set of reference panels built up of the SEA-specific genome dataset. We imputed a set of OA genotyping data and benchmarked it with the established reference panels namely the 1KGP phase 3 TOPMED, and harmonized 1KGP-Human Genome Diversity Project (HGDP). As such, incorporating OA and their rare variants, or SEA-specific alleles, in the reference panel has become crucial to support the notion that imputation is an integral part of unlocking more information about population genetics and disease studies, and a correct reference panel could be the key.
What were the main findings?
We cross-imputated publicly available datasets of Southeast Asia related ancestries from various resources, resulted in a final generated the SEA-specific reference panel dataset comprised of 2,550 samples with 113,851,450 variants.
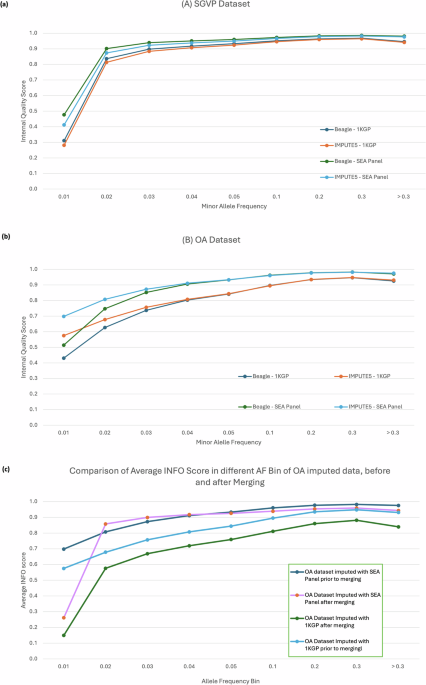
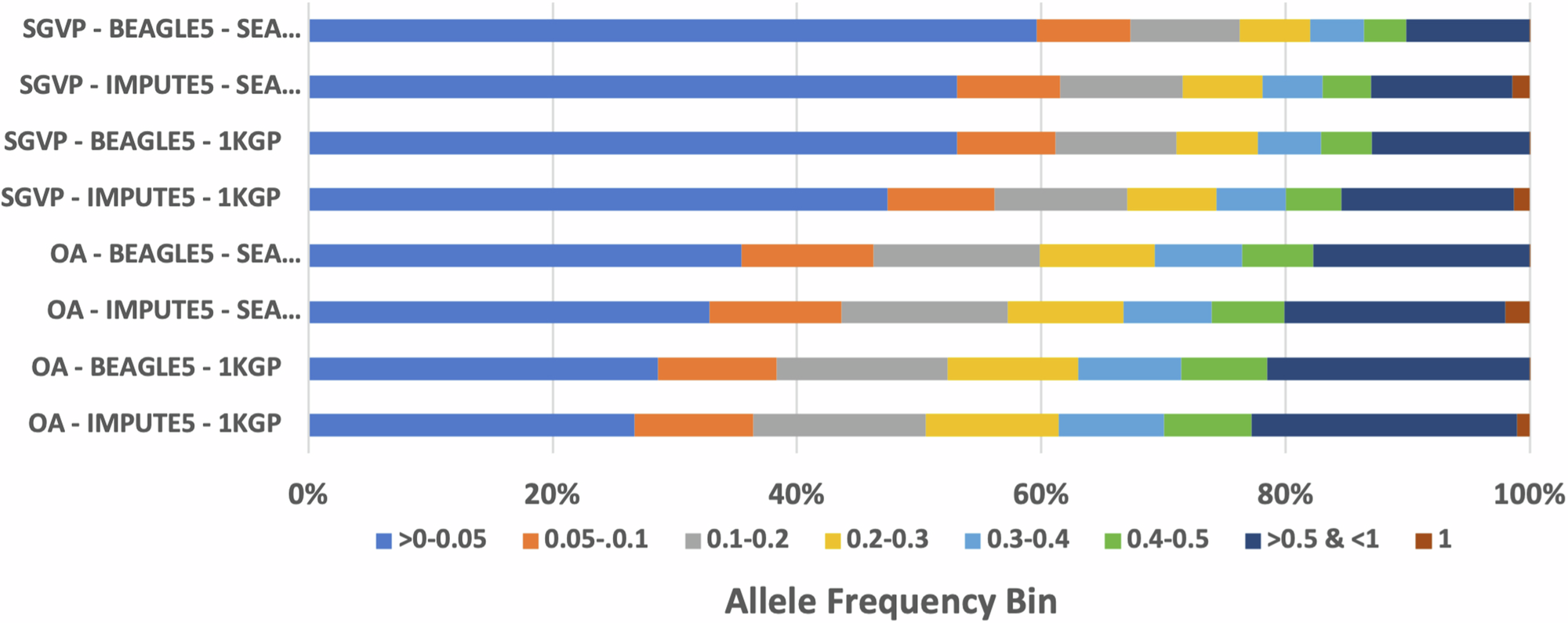
The SEA-specific panel produced more high confidence variants than 1000 Genomes Project (1KGP) when imputing the OA (8.9 million SEA-specific vs 8.1 million 1KGP) and the Singapore Genome Variation Project (SGVP) (12.5 million SEA-specific vs 11.8 million 1KGP) genotyping datasets.
We then compared the performance of our SEA panel with TOPMED and harmonized 1KGP-HGDP dataset. Both reference panels are larger in terms of sample size, with TOPMED R3 reaching 133,597 samples and 445,600,184 variants, and 1KGP-HGDP dataset with 4,091 samples and 75,917,155 variants after lift over from GRCh38 to GRCh37 to match our reference panel build and other reference panel. Our result shows that despite its large reference panel size, our SEA Panel imputed more high-quality polymorphic SNPs compared to both TOPMED and 1KGP-HGDP (as reflected by the number of SNPs with RSQ > 0.8), indicating ancestry closeness plays a more important role than the sample size of the reference panel.
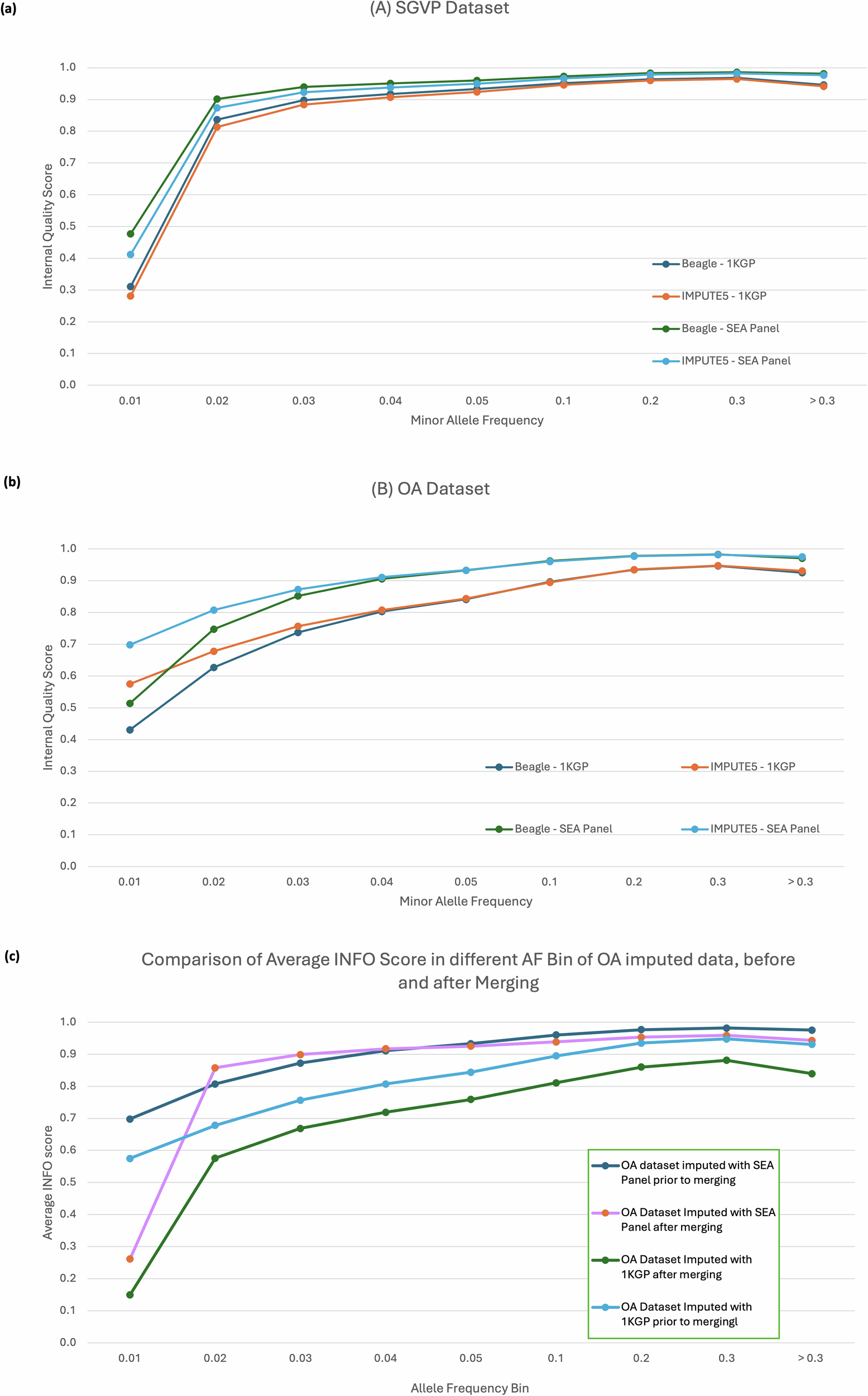
Imputation using the SEA-specific reference panel in the OA dataset yielded significantly more variants across all frequency bins when compared to the 1KGP panel. The biggest difference can be observed at lower frequencies, which yielded up to 50% more variants than the ones imputed with the 1KGP reference panel.
OA datasets imputed with SEA panels had a lower non-reference disconcordance rate by 3% and 10% higher recall to the original genotyping dataset, reaching ~886,000 variants recalled, not including initial SNP markers before imputation (~293,000 SNPs).
Conclusion:
We provided further supporting evidence that the choice of reference panel is an essential component in the imputation process, especially when studying underrepresented populations such as OA. The SEA-specific reference panel that we have developed, is expected to perform arguably better when imputing the Southeast Asian population, as demonstrated by the genotyping data of OA, Malays, and other SEA populations. Although the performance of the SEA-specific reference panel was simulated only with the SNP-array data, we believe that the imputation can be performed on the whole-genome sequencing datasets with comparable quality. On a separate note, we acknowledge, however, that the current imputation reference panel is limited by the number of available SEA representative datasets. The imputation accuracy may have been compromised as have been shown by lower disconcordance rate in TOPMED reference panel. More representative whole genome sequencing data of OA and other native populations from SEA would further increase the imputation power.
Research Interest:
- Molecular and Population Genetics; special interest on population genomics and evolution of the indigenous populations in Malaysia and Southeast Asia
- Genetic susceptibility of complex diseases, in particular infectious disease (dengue) and cardiovascular diseases; pharmacogenetics of hypertension
- Copy Number Variation (CNV)
"Ten Outstanding Young Malaysian 2013" under the category of Academic Achievement & Accomplishment.
Follow the Topic
-
npj Genomic Medicine
This is an international, peer-reviewed journal dedicated to publishing the most important scientific advances in all aspects of genomics and its application in the practice of medicine.

Related Collections
With Collections, you can get published faster and increase your visibility.
Diploid Genome and Disease
Publishing Model: Open Access
Deadline: Feb 07, 2027
Artificial Intelligence in Genomic Medicine
Publishing Model: Open Access
Deadline: Sep 23, 2026




Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in