A Graph Neural Network Model that Learns Polymer Properties
Published in Chemistry

Polyesters is a family of polymers composed of ester repeat units. The versatility of the esterification reaction allows many different types of multifunctional acids and glycols to be polymerized into polyesters and indeed some applications make use of multiple acids and multiple glycols in the same composition. Adding to this complexity are the various molecular weights and end-group distributions, as well as branching of the polymer backbone that have been realized. Together, this gives rise to an ever-increasingly large materials design space.
Computational property prediction tools, such as machine learning, allow chemists and material scientists to screen polymer combinations for desirable properties before embarking on expensive and time-demanding screening experiments. However, polymer property prediction is not straightforward for machine learning since it is challenging to represent polymers as machine-readable inputs. In this work, we seek to tackle these challenges.

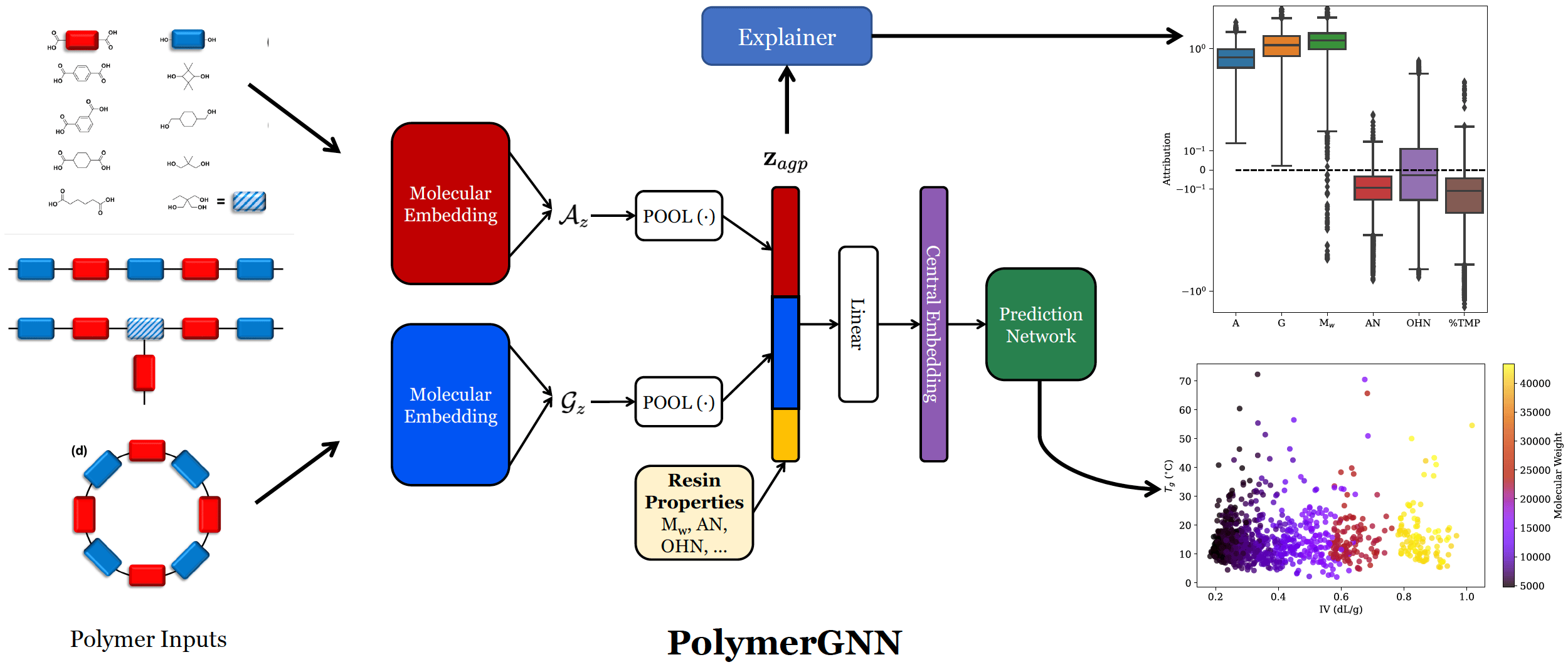
We propose PolymerGNN, a deep learning method to predict polymer properties. This model utilizes state-of-the-art graph neural networks (GNNs), which encode molecular graphs into fixed-length vectors on which the model can make predictions. GNNs perform iterative aggregation over edges within the graph, which are represented as chemical bonds, thereby leveraging atom-by-atom interactions to arrive at a rich molecular representation. Importantly, our method treats inputs as varying-size sets of monomers and then uses a type of deep learning - deep set learning - to learn properties on these sets. PolymerGNN also incorporates other vital experimental information such as molecular weight to produce robust predictions. We also develop a joint prediction architecture – training one model to learn multiple polymer properties at once. Our method demonstrates state-of-the-art results compared to previous machine learning algorithms, demonstrating its promise as a computational tool for polymer scientists.
We demonstrate the utility of PolymerGNN in a few ways. First, we present a new dataset with experimentally-validated measurements for Tg and IV on a set of diverse polymers synthesized by collaborators at Eastman Chemical Company. We utilize this dataset to train PolymerGNN, and we show that it can reliably predict both glass transition temperature (Tg) and intrinsic viscosity (IV). The prediction of Tg from monomer composition is straightforward and has been extensively explored in the past. On the contrary, IV depends on a variety of properties at both atomistic level and mesoscale, which makes the monomer-IV mapping a challenging task. Our joint prediction task has comparable performance to when PolymerGNN is trained to predict one property at a time, and it even achieves better performance on Tg prediction when trained jointly with IV. Next, we show how PolymerGNN can be used as a screening tool by predicting results on a randomly-generated library of polymers, which demonstrates how PolymerGNN is able to learn chemically-meaningful relationships. PolymerGNN produces predictions for one instance in under a second, allowing for high-throughput screens of thousands or even millions of combinations of polymers. Our model accounts for stoichiometry of the molecules, showing an ability to learn the varying properties of polymers when differing proportions of each monomer is used. Finally, we use interpretability techniques to peer into the behavior of PolymerGNN, and we find its behavior matches the intuition established by polymer science. Neural networks are infamously uninterpretable, causing great difficulties in understanding why a network made a given prediction. We utilize a gradient-based explainer to understand PolymerGNN’s predictions. This allows us to attribute predictions to different input variables, deriving a numerical score on which the importance of each variable can be compared.
We release PolymerGNN as an open-source codebase, inviting the community to utilize and build on top of our code. We hope that PolymerGNN can serve as a valuable tool for polymer property prediction and can inspire advances in the development of machine learning algorithms for polymer science. The full article can be accessed here: https://www.nature.com/articles/s41524-023-01034-3
Follow the Topic
-
npj Computational Materials
This journal publishes high-quality research papers that apply computational approaches for the design of new materials, and for enhancing our understanding of existing ones.

Related Collections
With Collections, you can get published faster and increase your visibility.
Altermagnetic Materials: Theory, Simulation, and Renewed Perspectives
Publishing Model: Open Access
Deadline: Feb 28, 2027
Recent Advances in Active Matter
Publishing Model: Open Access
Deadline: Sep 01, 2026

Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in