Molecular Fingerprints with Persistent Homology for Machine Learning Applications in Chemistry
Published in Chemistry


Machine learning applications for chemical problems are rapidly increasing the past few years. Their popularity is justified since they have led to the discovery of new molecules and materials with enhanced properties, new reactions, or have contributed to the reduction of computational effort needed of complex calculations and simulations. These are just a few examples about the success of data-driven approaches in chemistry but an open question that has not fully yet addressed is how a computational algorithm can efficiently “read” and “learn” patterns from molecular structures.
In this collaborative work between Jacob Townsend, John Hymel and Konstantinos Vogiatzis (Chemistry, University of Tennessee) and Cassie Micucci and Vasileios Maroulas (Mathematics, University of Tennessee), we are presenting a novel molecular representation method based on persistent homology, an applied branch of topology, which encodes the atomistic structure of molecules. Precisely, a molecule is mapped into a persistence diagram, a two-dimensional point summary, which demystifies the connected components and the empty space that exist in a molecule based on the atom types and the distances among them. A persistence diagram is further vectorized to a persistence image (PI), a weighted representation of the diagram, which captures the chemically driven uncertainty. The PI in that sense is a “molecular fingerprint”, and when used with machine learning, offers an efficient and reliable approach to screen large molecular databases when compared to other popular molecular representation schemes. The efficiency arises from the low computation effort needed to compare a large number of fingerprints, and the similar-size representations that are generated, independently of the molecular sizes.
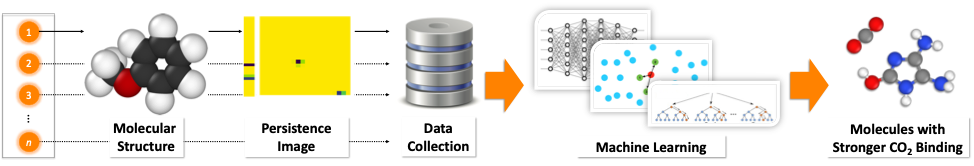
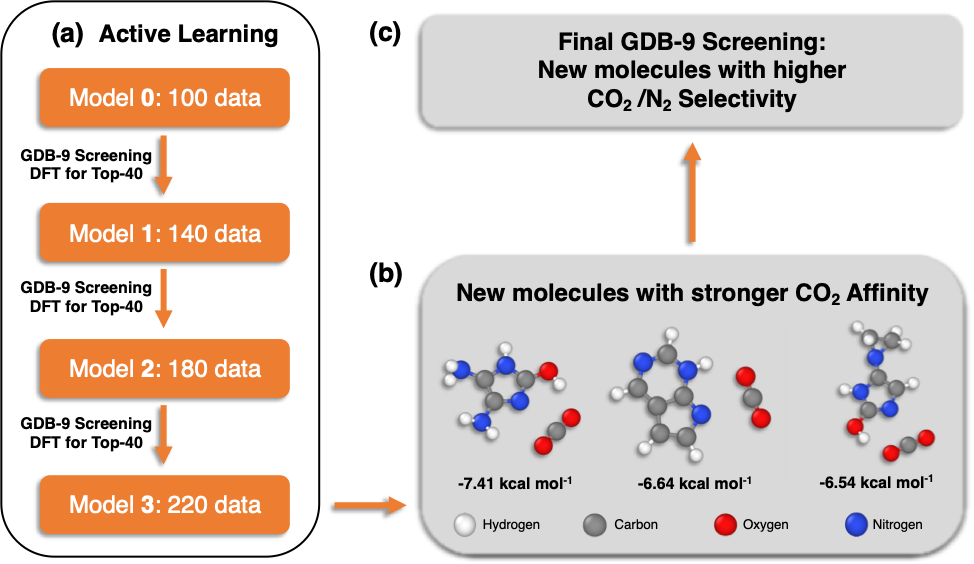
We demonstrated the applicability of the PI method by screening a large molecular database (GDB-9) with 133,885 organic molecules. Our target was to identify novel molecular units that selectively interact with CO2 and can be used as building blocks of materials, such as polymeric membranes. We began our study by computing with density functional theory (DFT) the CO2 interaction energies of 100 organic molecules. Since the initial, limited 100 data points were not capturing the diversity of the GDB-9 database, we applied a technique called active learning in order to incrementally obtain data which helped us efficiently screen the 133,885 molecules. We found out that the combination of PIs with active learning performed well with data (interaction energies) from only 220 molecules in order to identify new molecules with stronger CO2 binding. Finally, our data-driven methodology was able to identify molecular patterns previously unknown to us that increase the CO2 affinity of organic molecules.
The details of the PI molecular representation can be found on the article published in Nature Communications, 11, 3230.
Follow the Topic
-
Nature Communications
An open access, multidisciplinary journal dedicated to publishing high-quality research in all areas of the biological, health, physical, chemical and Earth sciences.

Related Collections
With Collections, you can get published faster and increase your visibility.
Women's Health
Publishing Model: Hybrid
Deadline: Ongoing
Tumor Microenvironment Crosstalk and Therapeutic Implications
Publishing Model: Hybrid
Deadline: Nov 02, 2026
Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in