Accelerating drug discovery through Open Access protein-ligand co-folding
Published in Protocols & Methods
Proteins interact with various molecules inside living bodies to carry out all the processes fundamental to sustaining life. Some examples of such molecules include other proteins, ligands, DNA, and RNA. Of particular interest to drug discovery are small molecules that bind to proteins and change their overall structure, as identifying and understanding these interactions allows us to design drugs that specifically target and modify proteins relevant to biomedical applications. Over the past decades, protein-ligand docking methods have been developed that find a suitable docking position for a given ligand on a given protein, often based on chemical interactions only within the selected binding site. While docking methods have been of tremendous use to the scientific community, they suffer from a vital issue: they usually require the holo structure of the protein. Holo structures are protein structures which already have a small molecule bound to them. Not only are these structures often not accessible for proteins which have yet to be targeted for drug development, but the use of holo structures does not take into account the flexibility of the proteins in the presence of small molecules, and limits the docking process to only known binding modes.
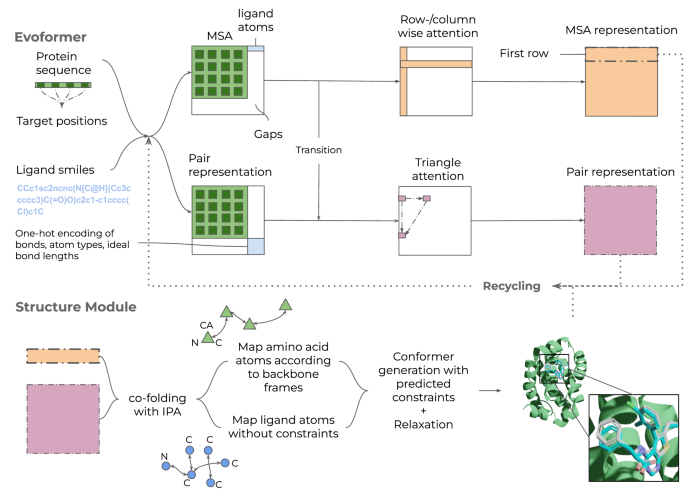
The advent of deep learning has heralded methods of “co-folding” protein-ligand complexes. Co-folding involves predicting the structure of the protein and ligand together rather than docking the ligand onto a holo structure. Thus, co-folding enables flexible modelling of proteins in the presence of ligands and can help in the discovery of new modes of binding, leading to improved structure prediction. Umol [1] (available at https://github.com/patrickbryant1/Umol), is an open-source co-folding method that takes as input a protein sequence, a ligand SMILES string, and information of the binding pocket, and predicts the structure of the protein and ligand, along with the plDDT, a confidence measure for the predicted structure. Owing to its innovative approach outlined below, Umol outperforms similar software in predicting the protein-ligand complexes.
Umol uses a modified EvoFormer module from AlphaFold2 [2], which processes protein sequence information in two tracks. First, a multiple sequence alignment (MSA) of the protein sequence, appended with ligand atoms, is constructed with a row- and column-wise self-attention. The MSA allows for identification of co-evolving residues and motifs and this information is passed to the pairwise representation track. Second, the pair representation track processes pairwise interactions both within the protein and between protein and ligand atoms using a triangle attention update mechanism. The ligand atoms are augmented to the pairwise representation of the protein using features like bonds and atom types.
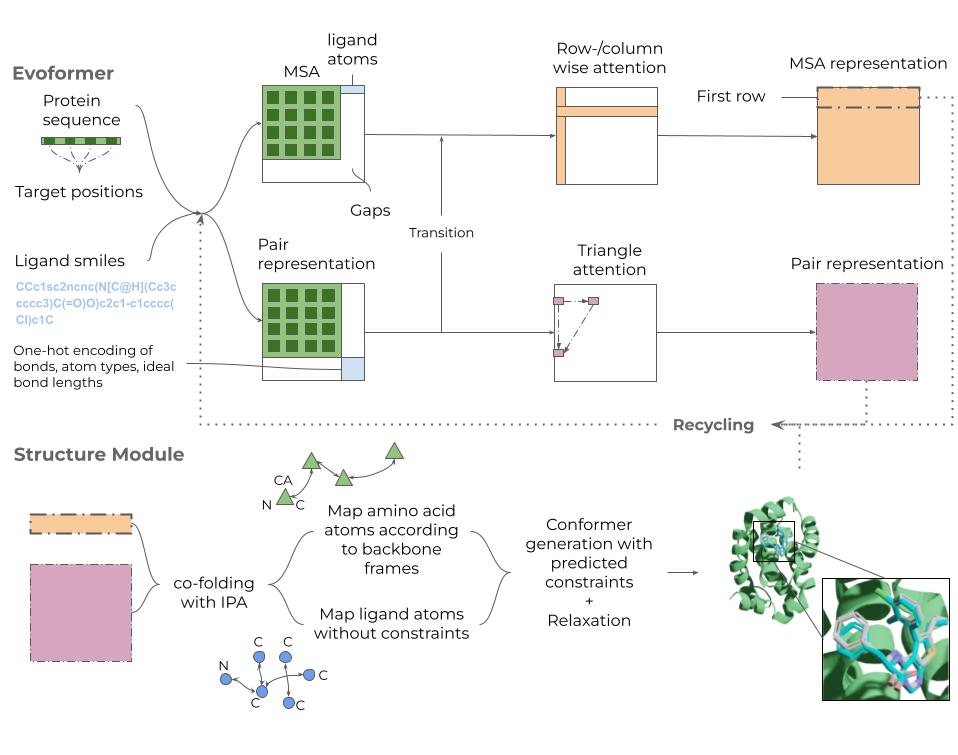
The EvoFormer module generates two outputs – an MSA representation for the individual protein residues and ligand atoms, and a pairwise representation of the protein-ligand complex. These outputs are passed to an Invariant Point Attention module which constructs the protein and ligand structure. To ensure that the predicted ligand structure is physically feasible, 100 conformers are generated for the ligand using RDKit (https://www.rdkit.org) and the structure closest to the atomic positions predicted by Umol is selected.
Umol also offers the option to run an energy relaxation of the predicted protein-ligand structure using a classical molecular force field. This step fixes the remaining atomistic clashes in the predicted structure.
To evaluate the success of our model, we compare the success rate (fraction of predictions with ligand RMSD ≤ 2 Å) of Umol with similar neural networks and find that Umol outperforms other methods in situations where the target structure is unknown. We also compare the similarity of the predicted protein-ligand structures (through ligand RMSD) to the confidence score assigned to a prediction (plDDT), for which we find a positive association. Finally, we assess whether Umol can correctly predict the binding affinity of ligands to their target protein (shown below) and find that the network can distinguish between strong and weak binders.
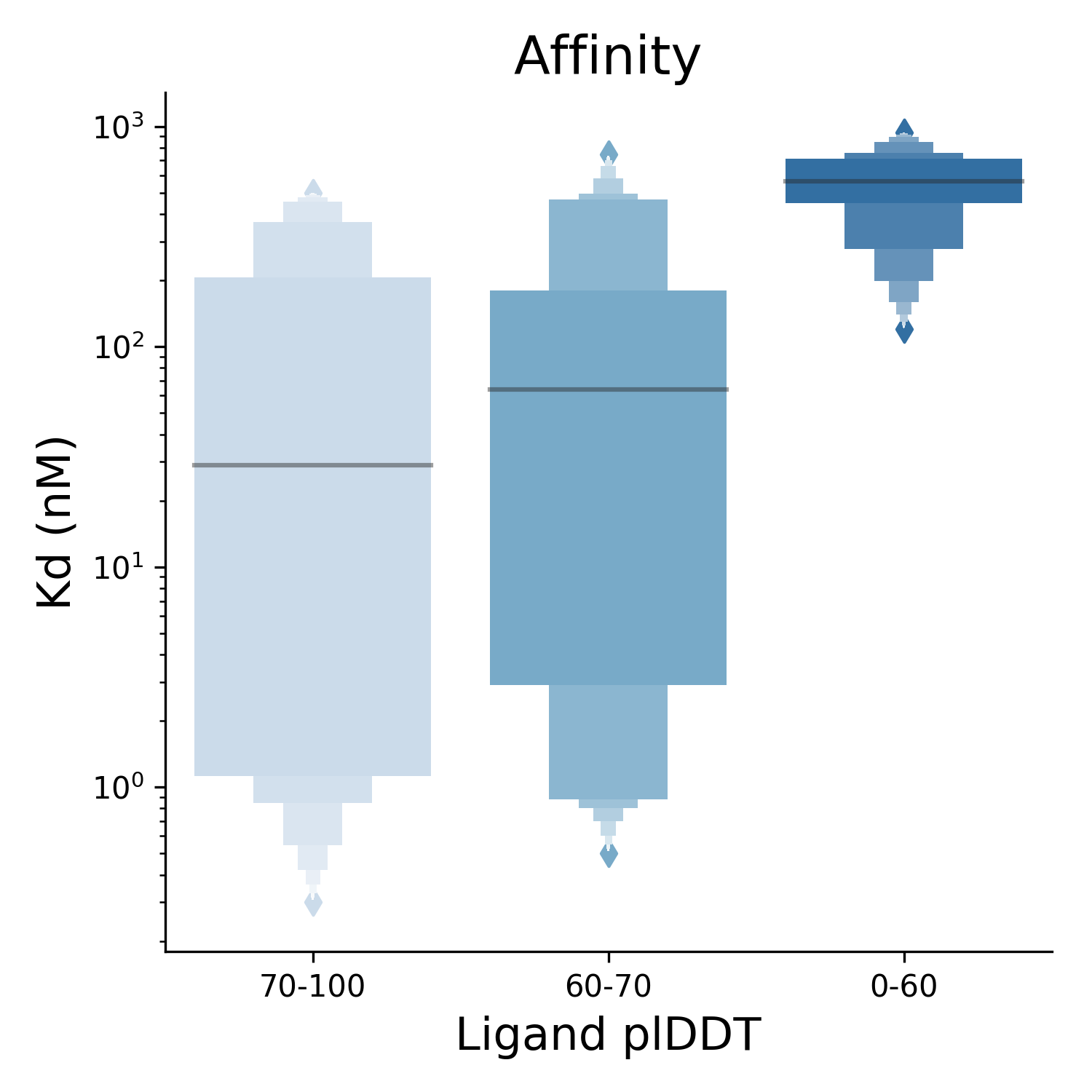 Protein-ligand affinity is an important measure of a drug molecule’s effectiveness in binding to its intended target. Using the predicted confidence from Umol, the plDDT, it is possible to distinguish high- and low-affinity binders. At ligand plDDT above 70, the median affinity is 30 nM, while below 60 plDDT, the median affinity is 500 nM. In addition, the plDDT is an effective estimate of the accuracy of the predicted complex.
Protein-ligand affinity is an important measure of a drug molecule’s effectiveness in binding to its intended target. Using the predicted confidence from Umol, the plDDT, it is possible to distinguish high- and low-affinity binders. At ligand plDDT above 70, the median affinity is 30 nM, while below 60 plDDT, the median affinity is 500 nM. In addition, the plDDT is an effective estimate of the accuracy of the predicted complex.
Open-access science has been critical to the progress of scientific research and innovation, especially within the machine learning ecosystem. Not only does this practice significantly enhance the accessibility of science, allowing limited-resource institutions to provide their scientists with high-quality tools, but it is also a step toward removing barriers between disciplines. Open sourcing enables researchers across different scientific fields to contribute their specific expertise to solve inter-disciplinary problems.
The framework presented in Umol builds on open-source innovations such as AlphaFold2 and other machine-learned approaches to biomolecular structure prediction. In keeping with this practice, Umol has been made publicly accessible on GitHub so that the scientific community may contribute insight and expertise for its further development. In addition, we expect that the success of the co-folding algorithm in overcoming the long-standing challenge of predicting protein-ligand complexes will have extensions to various scientific fields. While these potential uses include commonly referenced applications such as drug development, protein-ligand interactions are of primary interest in many fields including gene regulation, protein design, and immunology. We hope that in adhering to Open Access through the public release of Umol, we encourage its further development to tackle these interdisciplinary problems.
References
- Bryant, P., Kelkar, A., Guljas, A. et al. Structure prediction of protein-ligand complexes from sequence information with Umol. Nat Commun 15, 4536 (2024). https://doi.org/10.1038/s41467-024-48837-6
- Jumper, J., Evans, R., Pritzel, A. et al. Highly accurate protein structure prediction with AlphaFold. Nature 596, 583–589 (2021). https://doi.org/10.1038/s41586-021-03819-2
Follow the Topic
-
Nature Communications
An open access, multidisciplinary journal dedicated to publishing high-quality research in all areas of the biological, health, physical, chemical and Earth sciences.

Related Collections
With Collections, you can get published faster and increase your visibility.
Women's Health
Publishing Model: Hybrid
Deadline: Ongoing
Tumor Microenvironment Crosstalk and Therapeutic Implications
Publishing Model: Hybrid
Deadline: Nov 02, 2026


Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in