Accessible single-cell proteomics
Published in Protocols & Methods
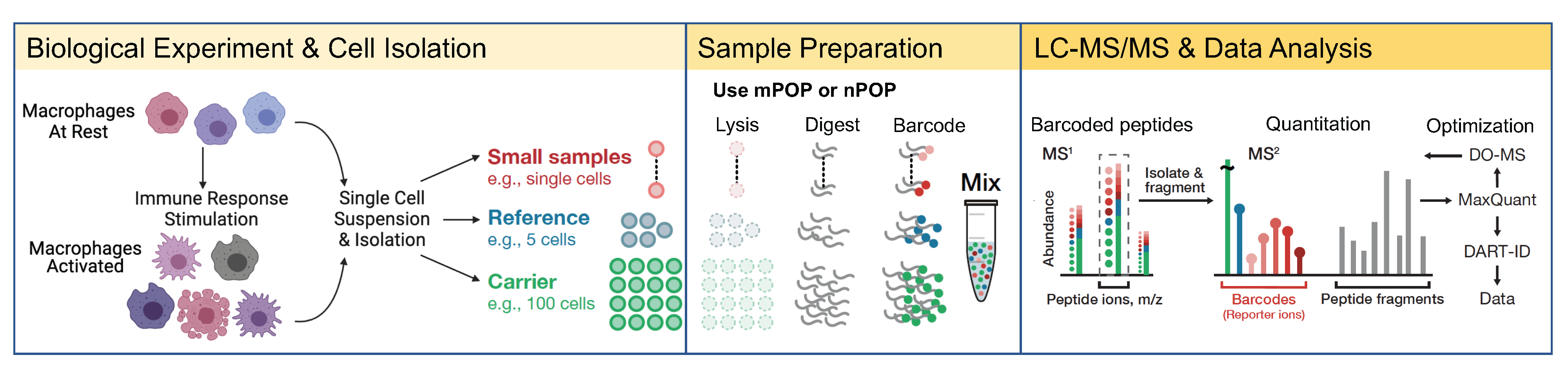

Recently single-cell mass-spectrometry analysis has allowed quantifying thousands of proteins in single mammalian cells. Yet, these technologies have been adopted in relatively few mass-spectrometry laboratories. Increasing their adoption can help reveal biochemical mechanisms that underpin health and disease, and it requires robust methods that can be widely deployed in any mass spectrometry laboratory.
This aim for a “model T” single-cell proteomics has been the guiding philosophy in the development of Single Cell ProtEomics by Mass Spectrometry (SCoPE-MS) and its version 2 (SCoPE2). We aimed to make every step easy to reproduce, from sample preparation and experimental parameters optimization to an open source data analysis pipeline. The emphasis has been on accuracy and accessibility, which has facilitated replication and adoption of SCoPE2. Yet, we still found that some groups adopting these single-cell technologies fail to make quantitatively accurate protein measurements, because they skip important quality control steps of sample preparation (such as negative controls and labeling efficiency), and mass spectrometry analysis, such as apex sampling and purity of MS2 spectra.
These observations motivated us to write a detailed protocol for multiplexed single-cell proteomics. The protocol emphasizes quality controls that are required for accurately quantifying protein abundance in single cells and scaling up the analysis to thousands of single cells. The protocol and its associated video and web resources should make single-cell proteomics accessible to the wider research community.
I received a BS from MIT in 2004 and then pursued doctoral research in the Botstein laboratory at Princeton University, aiming to understand how cells coordinate their growth, gene expression, and metabolism. As a postdoc in the van Oudenaarden laboratory at MIT, I characterized trade-offs of aerobic glycolysis (also known as Warburg effect) and obtained direct evidence for differential stoichiometry among core ribosomal proteins, suggesting that specialized ribosomes regulate protein synthesis.
My laboratory (slavovlab.net) aims to understand the rules governing emergent systems-level behavior and to use these rules to rationally engineer biological systems. We make quantitative measurements, often at the single-cell level, to test different conceptual frameworks and discriminate among different classes of models.
Areas of focus include:
• Ribosome mediated translational regulation: Mechanisms by which ribosomal modifications regulate protein synthesis
• Single-cell protein analysis: We develop and apply mass-spectrometry methods that allow analyzing thousands of proteins in single cells. These data have enabled characterizing post-transcriptional regulation in single cells at systems level, as well as discovering macrophage polarization in the absence of polarizing cytokines.
Follow the Topic
-
Nature Protocols
This journal publishes secondary research articles and covers new techniques and technologies, as well as established methods, used in all fields of the biological, chemical and clinical sciences.

Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in