One of the most important and well known physicochemical properties is the acidity constant, usually referred to as pKa and typically measured in (aqueous) solution. Predicting its value remains a challenge, which is addressed worldwide by very few research groups only. This is odd because commercial packages are known to sometimes fail badly or be it in need of further improvement when predictions are reasonable.
Over the last decade our lab perfected its own pKa prediction method, called AIBL, which is remarkably minimal yet powerful. In a way it is curious that this method has been overlooked during the development of the current spectrum of methods, which range from first principle calculations to de facto look-up tables. In fact, we stumbled on AIBL via a detour involving so-called quantum topological descriptors. The latter appear in a QSAR/QSPR approach called Quantum Topological Molecular Similarity (QTMS) (e.g. 1,2) that we developed long before AIBL. In QTMS the so-called bond critical points featured as the positions in a molecule at which various quantum chemical functions were evaluated to provide a “quantum fingerprint”3 of that molecule. The concept of molecular similarity grew even earlier from comparing whole electron densities or later electrostatic potentials. However, we proved that this approach was overkill and that bond critical properties sufficed as much more compact and effective descriptors.
Now, early on, we discovered the existence4 of local linear relationships between bond critical point properties and equilibrium bond lengths, only if the bonds vary little in their chemical surroundings. Such relationships break down completely, however, for larger subsets of bond critical points encompassing a wider variety of bonds. Hence, if one is interested in predicting pKa within a congeneric series of molecules, then bond lengths alone are enough. This realisation led to the birth of AIBL (pronounced “able”), which stands for Ab Initio Bond Length.
AIBL is an empirical-based method (i.e. needing calibration to experimental data), but one that employs quantum mechanically-derived 3D structural information as its descriptors. AIBL works on the basis that for a series of electronic congeners, an equilibrium bond length, typically in the gas phase and close to the site of ionisation, will display a linear relationship with aqueous pKa values. The Figure below illustrates the essence of AIBL in the context of tautomerisable compounds, a challenging set to predict pKa values for.
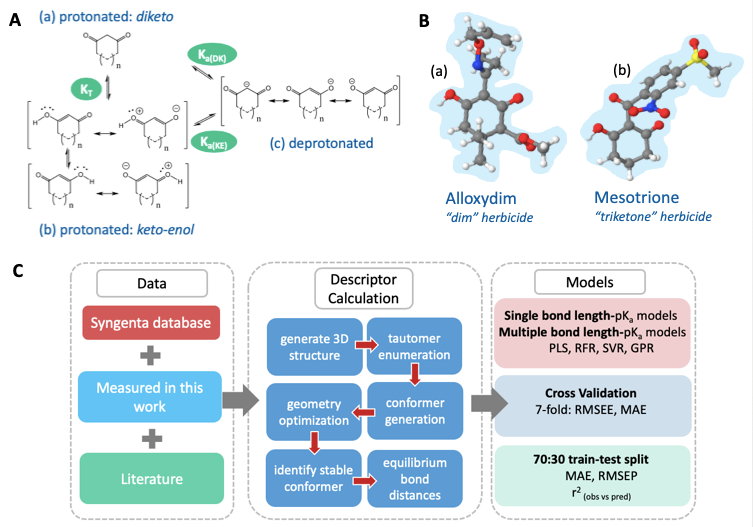
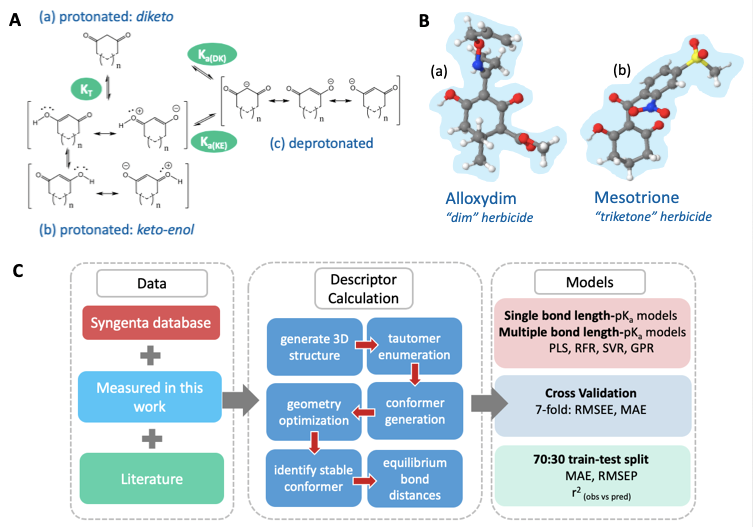
Figure A. (a) The diketo form of a 1,3-dione, (b) the resonance canonicals for the keto-enol form of 1,3-diones, and (c) the resonance canonicals for the anionic state, where n=0 or 1 if the ring is five- or six-membered, respectively. KT denotes the equilibrium constant between tautomeric states, Ka(DK) denotes the dissociation equilibrium from the diketo state and Ka(KE) the dissociation equilibrium from the keto-enol state. B. (a) The global minimum geometry of Alloxydim, a 2-oxime herbicide and Mesotrione in the keto-enol anti state, (b) a triketone herbicide. C. The AIBL-pKa workflow implemented here for cyclic -diketones.
Since our original proof-of-concept study on phenols5, we have proven the existence of this simple relationship between solution pKa and equilibrium bond length(s) for a variety of chemistries: benzoic acids6, anilines6, naphthols7, phenylacetic, benzohydroxamic and phenoxyacetic acids8, bicyclo[2,2,2]octane and cubane carboxylic acids9, aryl guanidines and 2-(arylamino)imidazolidines10, guanidines11, guanidine-containing compounds12, sulphonamide drugs13 and tautomerisable herbicides involving 1,3-cyclopentanediones and 1,3-cyclohexanediones14 and other case studies yet to be published, including amidines.
AIBL shows particularly impressive accuracy compared to Marvin, a commercial program by ChemAxon used to predict pKa values for every drug on the online database “DrugBank”: an MAE of 0.20 for a series of 27 2-(arylimino)imidazolidine drugs. The significance of these results is that these compounds contain the guanidine functional group, which is known to be notoriously difficult to predict for. Our work also demonstrates accurate predictions for multiprotic compounds, and most rewardingly, correction of erroneously measured pKa values.
A theoretician's dream is to truly predict an experimental value. This happened on a number of occasions but most spectacularly while collaborating with Dr Christophe Dardonville in Madrid, Spain, who uses state-of-the-art equipment to measure pKa values. Confident in the predictive capacity of AIBL we told him before his measurement that the answer for the drug Clozapine was 7.76, and teased him in an email asking why one still needed to measure it. Christophe’s returning E-mail stated that the experimental result was 7.77 and that, as a previous non-believer, he was now converted.
The AIBL method has several advantages over other techniques:
(1) Low energy conformations must be identified for 4 species for thermodynamic cycle-based methods, yet AIBL requires only 1, thus vastly reducing computation time. Calculations still take longer than purely empirical methods but there is often a pay-off in terms of increased accuracy.
(2) High levels of theory are required to derive accurate Gibbs energies of the species involved in the pure thermodynamic cycle method. AIBL can work at HF/6-31G(d) but currently B3LYP/6-311G(d,p) is recommended given reasonable timescales.
(3) The relationship between the descriptor and observable is not a “black box”. This means that the occurrence of outliers can usually be explained in a physically meaningful way, i.e. in terms of geometric or chemical incongruence.
(4) Multiprotic compounds can be predicted for (proven yet again for sulfonamides and sulfanilamides).
(5) We have developed workflows to form predictive models for tautomerisable compounds including guanidines, amidines, triketones and diketones. We expect this to be applicable to other instances of tautomerisation.
(6) The use of QM-derived 3D structures means that hydrogen bonding and steric effects on pKa are implicitly accounted for, without the need for parameterisation, as in the case for some 2D methods.
(7) Predictions for compounds with ortho-substituents can be problematic due to intramolecular interactions (i.e. hydrogen bonding) with the ipso ionisable group. Our method is capable of making predictions for ortho-compounds by formation of specific predictive equations for subsets of compounds featuring each type of interaction. A meaningful subset can involve just 5 compounds.
In summary, AIBL has proven to be so reliable that if one finds an outlier in its one of its predictions then the only possibilities are that (i) the experiment is wrong and needs to be redone, (ii) an error was made in the bond length calculation, or (iii) the data point belongs to a new subset of congeners that needs to be calibrated. That AIBL would not work is simply not an option… With this happy observation in mind, we hope that researchers will start using AIBL when reliability is important or when measurements are inconsistent or missing.
References
1. O'Brien, S.E. & Popelier, P.L.A. Quantum Molecular Similarity. Part 3 : QTMS descriptors. J.Chem.Inf.Comput.Sci. 41, 764-775 (2001).
2. Popelier, P.L.A. & Smith, P.J. QSAR models based on Quantum Topological Molecular Similarity. Eur.J.Med.Chem. 41, 862-873 (2006).
3. Popelier, P.L.A. Quantum molecular similarity. 1. BCP space. J.Phys.Chem.A 103, 2883-2890 (1999).
4. O'Brien, S.E. & Popelier, P.L.A. Quantum molecular similarity. Part 2: The relation between properties in BCP space and bond length. Can.J.Chem. 77, 28-36 (1999).
5. Harding, A.P. & Popelier, P.L.A. pKa Prediction from an ab initio bond length: Part 2—phenols. Phys.Chem.Chem.Phys. 13, 11264–11282 (2011).
6. Harding, A.P. & Popelier, P.L.A. pKa prediction from an ab initio bond length: Part 3—benzoic acids and anilines. Phys.Chem.Chem.Phys. 13, 11283–11293 (2011).
7. Anstöter, C., Caine, B.A. & Popelier, P.L.A. The AIBLHiCoS Method: Predicting Aqueous pKa Values from Gas-Phase Equilibrium Bond Lengths. J.Chem.Inf.Model. 56, 471−483 (2016).
8. Alkorta, I. & Popelier, P.L.A. Linear free energy relationships between a single gas-phase ab initio equilibrium bond length and experimental pKa values in aqueous solution. ChemPhysChem 16, 465-469 (2015).
9. Alkorta, I., Griffiths, M.Z. & Popelier, P.L.A. Relationship between experimental pKa values in aqueous solution and a gas phase bond length in bicyclo[2.2.2]octane and cubane carboxylic acids. J.Phys.Org.Chem. 26, 791-796 (2013).
10. Dardonville, C., et al. Substituent effects on the basicity (pKa) of aryl guanidines and 2-(arylimino)imidazolidines: correlations of pH-metric and UV-metric values with predictions from gas-phase ab initio bond lengths. New J.Chem. 41, 11016-11028 (2017).
11. Griffiths, M.Z., Alkorta, I. & Popelier, P.L.A. Predicting pKa values in aqueous solution for the guanidine functional group from gas phase ab initio bond lengths. Mol.Inf. 32, 363-376 (2013).
12. Caine, B.A., Dardonville, C. & Popelier, P.L.A. Prediction of Aqueous pKa Values for Guanidine-Containing Compounds Using Ab Initio Gas-Phase Equilibrium Bond Lengths. ACS Omega 3, 3835−3850 (2018).
13. Caine, B.A., Bronzato, M. & Popelier, Paul L.A. Experiment stands corrected: accurate prediction of the aqueous pKa values of sulfonamide drugs using equilibrium bond lengths. Chem.Sci. 10, 6368-6381 (2019).
14. Caine, B.A., et al. Aqueous pKa prediction for tautomerizable compounds using equilibrium bond lengths. Comm.Chem. 3, 21 (2020).
Follow the Topic
-
Communications Chemistry
An open access journal from Nature Portfolio publishing high-quality research, reviews and commentary in all areas of the chemical sciences.

Related Collections
With Collections, you can get published faster and increase your visibility.
Chemical modification of proteins
Publishing Model: Open Access
Deadline: Sep 30, 2026
Sustainable waste management through polymer upcycling
Publishing Model: Open Access
Deadline: Aug 31, 2026


Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in