An embryonic protein and an old drug in the spotlight of aggressive KMT2A-rearranged leukaemia
Published in Cancer
Explore the Research

SET-PP2A complex as a new therapeutic target in KMT2A (MLL) rearranged AML - Oncogene
Oncogene - SET-PP2A complex as a new therapeutic target in KMT2A (MLL) rearranged AML
Cancer is a genetic disease. This means that it is due to mutations in the genes that control how cells grow and multiply. As our body gets old, it makes more and more mistakes in its daily tasks, including replicating the DNA, the molecule that contains the code to govern all the activities and the fate of our cells. This model explains why cancer affects middle-age people, with median age at diagnosis of 60-65, as it takes some years to accumulate the DNA mutations that eventually determine the development of cancer.
Wait a minute.
Children also get cancer. Blood cancers, known as leukaemia, are the most commonly diagnosed cancers in children and among people in their 20s. A particularly aggressive form of leukaemia is KMT2A-rearranged (R) leukaemia, also called MLL or mixed lineage leukaemia, which is due to mutations affecting the gene KMT2A.
If it takes less time for someone with this mutation to develop leukaemia, does this mean that some genetic mutations, like this one, are more “potent” than others?
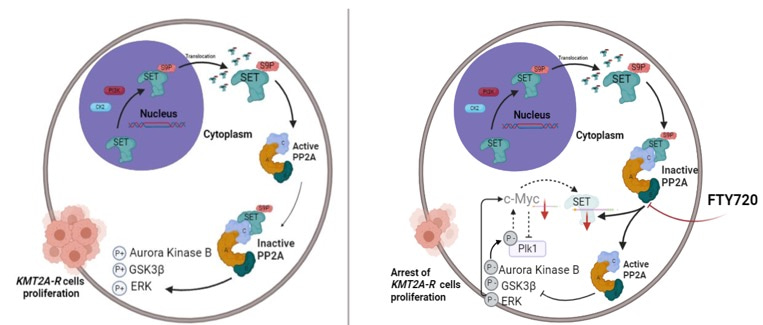
Yes, indeed. Many scientists working in the field of KMT2A-R leukaemia have dedicated their careers to understanding how the proteins encoded by KMT2A mutations, specifically by the KMT2A chromosomal translocations, work, with the hope to find a way to “switch them-off” and cure the patients affected by this leukaemia. We have learnt that KMT2A chromosomal translocations give instructions to progenitor blood cells to express genes that, normally, would be expressed only in embryonic cells; these embryonic genes are silenced in “grown-up” blood cells of healthy people. Among these genes they found one called SET, that encodes for a little protein that shuttles between different compartments within a cell.
What does SET do?
I was waiting for this question! SET is an inhibitor of a protein, the phosphatase PP2A, that is a master regulator of the growth and survival of cancer cells. The phosphatase PP2A can control cellular growth and survival by modulating the activity of several signalling pathways, acting as a sort of brake. But if SET accumulates within the cells, the activity of PP2A is inhibited. When the brake does not work, cellular growth accelerates out of control.
Is SET expressed only in patients with KMT2A-R leukaemia? What does it do to leukemic cells? And, what happens if we switch off the expression of SET in leukemic cells?
To answer these questions, we used bioinformatic approaches and we analysed the expression of SET in large datasets of acute myeloid leukaemia (AML) patients. This analysis revealed that the expression of SET correlates with poor prognosis, meaning that the patients with high level of SET expression have smaller chances to survive. We also found that SET is abundantly expressed in all the AML subtypes, not just those with KMT2A translocations. By using other bioinformatic approaches we discovered that there is a very strong correlation between SET and the expression of other genes, called HOXA genes and MEIS, that have been known for a long time as genes critically activated by KMT2A proteins to convert progenitor blood cells in leukemic cells, a process known as “leukemogenesis”.
So, what happens if we switch off the expression of SET in leukemic cells?
I can’t wait to tell you! By using a technique called RNA interference, we knocked down SET in KMT2A-R leukemic cells and then we analysed their gene expression profile. We discovered that when SET is knocked down, the expression of genes HOXA and MEIS is reduced. More importantly, these cells are unable to grow and form colonies in vitro! This, overall, means that SET is important for the growth and survival of KMT2A-R leukemic cells and it is also important to maintain the expression of some of the key genes including HOXA and MEIS, that maintain the leukemogenic programme in the KMT2A-R leukemic cells.
Cool, does this mean that by targeting SET we could kill the KMT2A-R leukemic cells and cure the patients? Is there any drug that we can test?
There is a drug called FTY720 (Fingolimod), already approved by the Food and Drug Administration for patients affected by multiple sclerosis. It turns out that FTY720 can inhibit SET by interfering with its ability to bind the protein phosphatase PP2A. This drug had been already tested in some pre-clinical models of leukaemia and proved effective. We tested it in our models, and we discovered that it halted the growth of KMT2A-R leukemic cells and made them more responsive to standard chemotherapy. This is important as KMT2A-R leukaemia are often resistant to standard chemotherapy-based treatment. To understand the molecular consequences of FTY720 treatment in KMT2A-R leukemic cells and the signalling pathways targeted, we used phospho-proteomics. The analysis revealed that FTY720 affected signalling pathways important for cell division, cell death and gene expression. To understand which were the genes whose expression changed upon FTY720 treatment, we used an approach called RNA-seq. This technique allowed us to analyse the whole gene expression profile and to discover that the expression of many genes identified as important in the leukemogenesis driven by KMT2A, as HOXA and MEIS, as well as MYC, another gene important for the proliferation of KMT2A-R leukemic cells, was reduced when the leukemic cells were treated with FTY720. Our data indicate that this is due to the effect of FTY720 on various signalling pathways.
So, can we use FTY720 to kill KMT2A-R leukemic cells and cure the patients?
This is unfortunately unlikely. FTY720 is an immune suppressor, therefore it might debilitate the already compromised immune system of leukemic patients. But as our data indicate that SET is so important for the survival of KMT2A-R leukemic cells, we could use other strategies to knock down or inhibit SET and we can test the non -immunosuppressive analogues of FTY720 that are already available. I can’t wait to get back to the lab to continue to work on this project! Wait…I don’t have the funding! Let me get back to my office on that grant proposal!
Dr. Maria Teresa Esposito is a Senior Lecturer in Biochemistry at the University of Surrey, UK. Maria Teresa completed her PhD in Molecular Medicine in Napoli, Italy, in 2009 and then she took a post-doctoral training in haemato-oncology at King’s College London, where she studied the molecular mechanisms required for establishment and maintenance of acute myeloid leukaemia (AML) stem cells. In 2017 she was awarded the Leukaemia UK John Goldman Fellowship for translational haematology and started her independent research focusing on exploring and exploiting protein phosphorylation for the identification of new therapies for MLL (KMT2A) leukaemia. Maria Teresa is passionate about education, inspiring the next generation of scientists and science professionals, and public engagement.
Follow the Topic
-
Oncogene
This journal aims to make substantial advances in our knowledge of processes that contribute to cancer by publishing outstanding research.





Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in