An end-to-end workflow to study newly synthesized mRNA following rapid protein depletion in Saccharomyces cerevisiae
Published in Chemistry, Protocols & Methods, and Cell & Molecular Biology
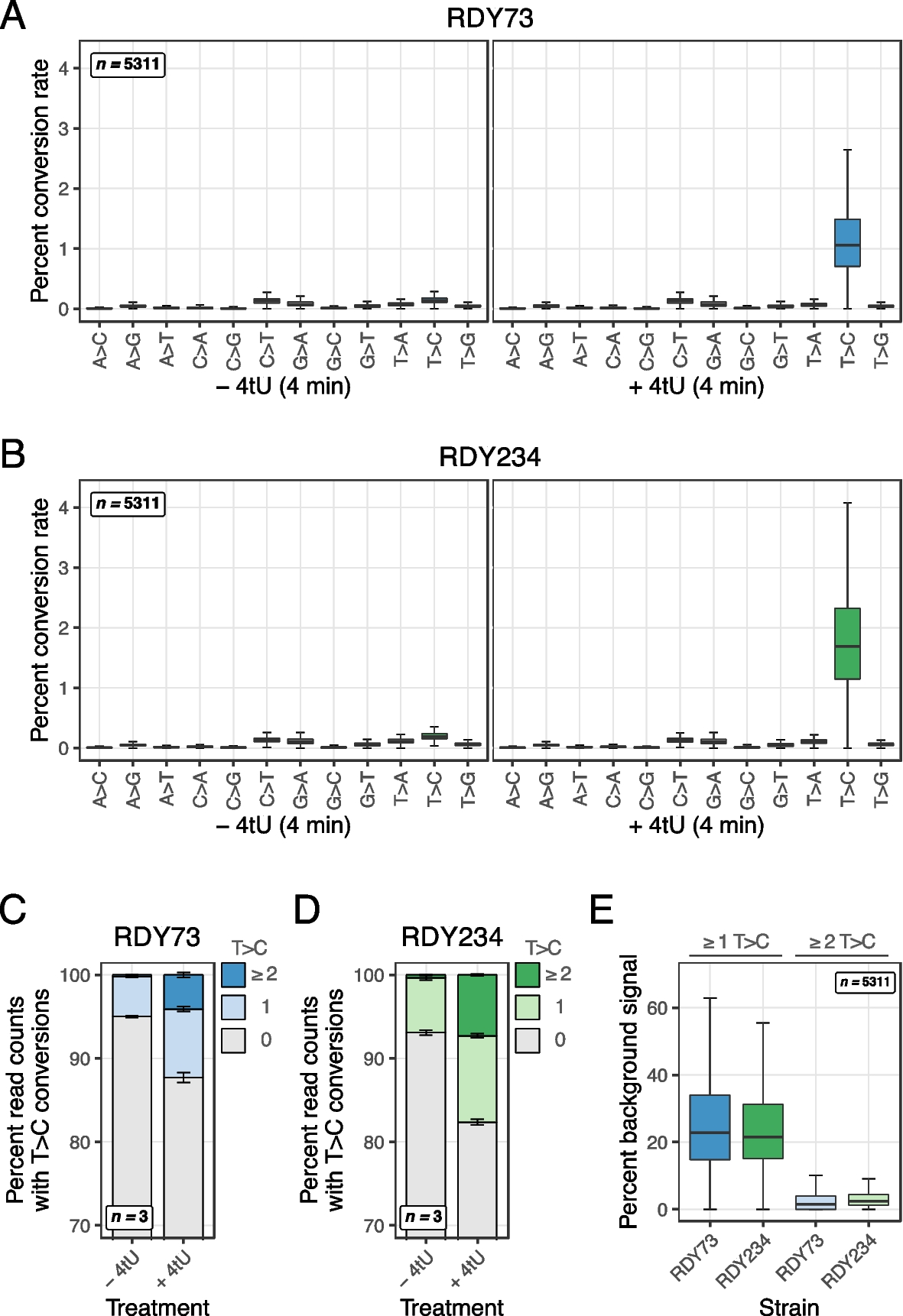

Join Gabriel Gasque, Head of Outreach at protocols.io, as he interviews John Ridenour and Rafal Donczew, the authors of a recently published protocol in BMC Methods, presenting an end-to-end workflow to deplete proteins of interest and measure newly synthesized RNA in Saccharomyces cerevisiae.
Get ready for exclusive insights, behind-the-scenes secrets, and a glimpse into the future of their innovative work.
Chiara is a Senior Editor for the BMC Series and joined in April 2022 after over ten years of research experience. She holds a Master’s degree in Medical Biotechnologies and a PhD in Neuroscience from the University of Milano-Bicocca, where she studied the effect of targeted cell cycle inhibition on glioma stem cells. Following postdoctoral work at the same University, she moved as a Research Fellow to the University of Sussex where she focused on the identification of potential new biomarkers and therapeutic targets in glioblastoma and breast cancer.
Follow the Topic
-
BMC Methods
An open-access, peer-reviewed journal that focuses on publishing lab protocols and methodology papers in the natural sciences; including biology, chemistry, physics, computational and biomedical sciences.

Related Collections
With Collections, you can get published faster and increase your visibility.
Cell communication analysis technology
Cell communication is a fundamental process that underpins numerous biological functions, from immune responses to tissue repair. The intricate signaling pathways that govern how cells communicate are crucial for maintaining homeostasis and regulating physiological responses. Recent technological advances, including single-cell RNA sequencing, mass cytometry, and high-resolution imaging, have dramatically improved our ability to study these networks and understand how cells coordinate their behavior in response to external cues. Computational tools can now infer intercellular signaling from single-cell and spatial transcriptomics data, identifying key ligand-receptor pairs and signaling pathways. Spatially informed approaches further map these interactions with high resolution, revealing how a cell’s location shapes its communication with neighbors. Complementing these computational methods, experimental technologies such as genetic marking systems can directly label contacting cells in vivo, allowing researchers to track interaction history and study how cellular crosstalk drives development and disease progression.
Future advancements may include the integration of artificial intelligence and machine learning to analyze complex datasets, the creation of real-time monitoring systems for live-cell interactions, and the establishment of standardized protocols for cell communication analysis. These innovations could significantly enhance our ability to dissect cellular communication networks and their roles in health and disease.
In light of these developments, BMC Methods is opening a collection on “Cell communication analysis technology.” Topics of interest include:
- Novel imaging techniques for cell communication
- Advancements or improvements in single-cell analysis methods
- Computational modeling of cell signaling pathways
- Intercellular communication in cancer progression
Submissions should present an experimental or computational method for the analysis of cell–cell communication. The method may be completely new or offer an improved version of an existing method, but in either case it must represent a clear advance over what is currently available, be tested and validated, and include a discussion of its advantages and limitations relative to alternative approaches. Protocols describing routine or well-established methods without methodological innovation will not be considered for publication.
All manuscripts submitted to this journal, including those submitted to collections and special issues, are assessed in line with our editorial policies and the journal’s peer review process. Reviewers and editors are required to declare competing interests and can be excluded from the peer review process if a competing interest exists.
Publishing Model: Open Access
Deadline: Nov 09, 2026
Optogenetics and photopharmacology: precision control of biological systems
The past two decades have seen remarkable advances in our ability to control and interrogate biological systems with light. Optogenetics and photopharmacology have emerged as two complementary disciplines that harness the precision of light to modulate molecular, cellular, and physiological processes with unprecedented spatiotemporal resolution.
Optogenetics, which broadly refers to the genetic introduction of light-sensitive proteins into specific cells or tissues, allows for non-invasive, reversible control of cellular activity. Since its inception, optogenetics has transformed neuroscience by enabling targeted activation or silencing of neurons with millisecond precision. However, its applications have rapidly expanded beyond the nervous system to include studies in cardiac physiology, developmental biology, immunology, and even synthetic biology. From light-gated ion channels and pumps to engineered transcription factors and signal transducers, optogenetic tools now offer a diverse array of functional capabilities.
On the other hand, photopharmacology involves the use of light-activated drugs to manipulate biological pathways, offering a way to study cellular mechanisms and therapeutic interventions. Unlike traditional pharmacological agents, these photoresponsive compounds offer reversible, localized control over protein function without the need for genetic manipulation. By designing ligands that switch between active and inactive states under different wavelengths, researchers can modulate enzyme activity, receptor function, or protein-protein interactions with high specificity. This approach has significant advantages in contexts where genetic access is limited or where reversible, rapid modulation of endogenous targets is desired.
Together, these techniques provide an important way of dissecting complex biological systems and enhancing our understanding of cellular functions. Despite their transformative potential, implementing optogenetic or photopharmacological methods in the laboratory requires careful planning, technical expertise, and an understanding of both the biological system and the optical tools involved. Light delivery, gene expression, molecular targeting, and real-time analysis all demand fine-tuning for successful outcomes.
BMC Methods is opening this Collection on “Optogenetics and photopharmacology: precision control of biological systems” to further consider the nuances of these techniques. Topics of interest include but are not limited to:
Advances in light-sensitive proteins for optogenetics
Light delivery systems and setup for in vivo and in vitro experiments
Genetic tools for optogenetic manipulation
Chemical design and application of photoswitchable ligands
Tissue- and cell-specific targeting strategies
Real-time imaging techniques and optical readouts
All manuscripts submitted to this journal, including those submitted to collections and special issues, are assessed in line with our editorial policies and the journal’s peer review process. Reviewers and editors are required to declare competing interests and can be excluded from the peer review process if a competing interest exists.
Publishing Model: Open Access
Deadline: Sep 07, 2026

Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in