Approaches to the molecular pharmacology of PI3Kδ
Published in Cancer


In our article we describe
- the generation of a PI3K isoform-selective mechanistic cell line model,
- the I777M mutation in p110δ, which markedly reduces sensitivity to idelalisib,
- comparative high-throughput assessment of currently developed PI3K inhibitors,
- structural investigation of the effects of the I777M mutation on PI3K inhibitor binding.
These four areas build on previous findings that we will refer to in this blog post via commentaries rather than the original work.
BaF3 cells for functional kinase screens
The pro-B cell line BaF3 is useful for physiologically relevant kinase screens, for instance massively parallel active tyrosine kinase-dependent cellular assays 1. By generating isogenic PI3K isoform-dependent BaF3 cells, we extend this approach to the lipid kinases p110δ and p110α. While tyrosine kinases, e.g. the non-oncogenic, B cell receptor (BCR)-associated kinase BTK, require activation by gene fusions 2, the C-terminal point mutation E1021K together with overexpression is sufficient to achieve p110δ-dependent growth of BaF3 cells. The newly developed cell line model offers sensitive PI3K isoform-selective inhibitor assessment simply by monitoring cell growth. Avoiding issues with membrane-resident reactants and preparations of isolated enzymes, this system more faithfully and coherently reflects pre-clinical efficacies than biochemically determined PI3K inhibitor potencies and has advantages also over existing cellular systems that monitor Akt phosphorylation.
Chemical genetics: unnatural ligands and mutant enzymes
The development of ATP binding site-directed kinase inhibitors has resulted in potent anti-cancer drugs. The interaction of tyrosine kinases with such inhibitor molecules is partly determined by the size of the so-called gatekeeper residue, which gives access to a portion of the kinase active site that is not used by ATP. Somatic mutation at this residue has direct medical relevance, when cancer cells escape selection pressure imposed by clinically applied inhibitors 3. The same mechanism applies for acquired resistance to the tyrosine kinase inhibitor ibrutinib, which can be caused by mutation of cysteine 481, where ibrutinib covalently binds 2. In the laboratory, engineered resistance mutations can be used for a chemical genetic strategy to relate tyrosine kinase activity with involvement in cell functions, leading e.g. to the detection of functional dasatinib targets 4. These discoveries in the tyrosine kinase field inspired our investigation of PI3K isoforms.
In the binding pocket of the lipid kinase p110α, the I800M mutation was identified as a sensitivity hotspot different from the gatekeeper residue, raising the expectation that it might cause acquired resistance to PI3K inhibitors 5. This prompted us to investigate, whether the analogous I777M mutation in the PI3K isoform p110δ affects sensitivity to idelalisib, the first clinically applied PI3K inhibitor and an efficient, p110δ-selective anti-lymphoma drug. Indeed, p110δ-I777M maintains cellular phospho-AKT levels at approximately 100-fold higher idelalisib concentrations than the wild type kinase. Employing this mutant kinase and idelalisib as a newly developed chemical genetic tool set, we demonstrate engagement of p110δ in BCR-triggered cytokine secretion, chemotaxis to CXCL12 and p110δ-dependent cell growth. Despite this evidence of the functional capacity of the I777M mutation to reconstitute physiologically relevant cell functions in the presence of idelalisib, it has become obvious in the meantime that clinically observed idelalisib resistance is not due to mutations in p110δ or the PI3K/ATK pathway altogether, but based on compensatory mechanisms 6. One could speculate that this striking difference to tyrosine kinases may be explained by differently controlled signal transduction via membrane-resident second messengers as opposed to protein phosphorylation cascades. Regardless of its lack of clinical relevance, the I777M mutation in p110δ can be useful in drug development cycles involving chemical and genetic modifications.
Functional assessment of PI3K inhibitors
Extensive drug development efforts in the PI3K inhibitor field during the last two decades necessitate commensurate pre-clinical assessment 7. To this end, we applied our newly generated cellular model system with the capability to dissect the isoform selectivity of these substances for a detailed pharmacological analysis in a concentration-response format. Quantitative comparisons of 33 PI3K inhibitors in a coherent system yielded a superior reference resource (Figure 1). The scale of this endeavor demanded parallel cultures in 384-well plates and a robotic pipetting system.
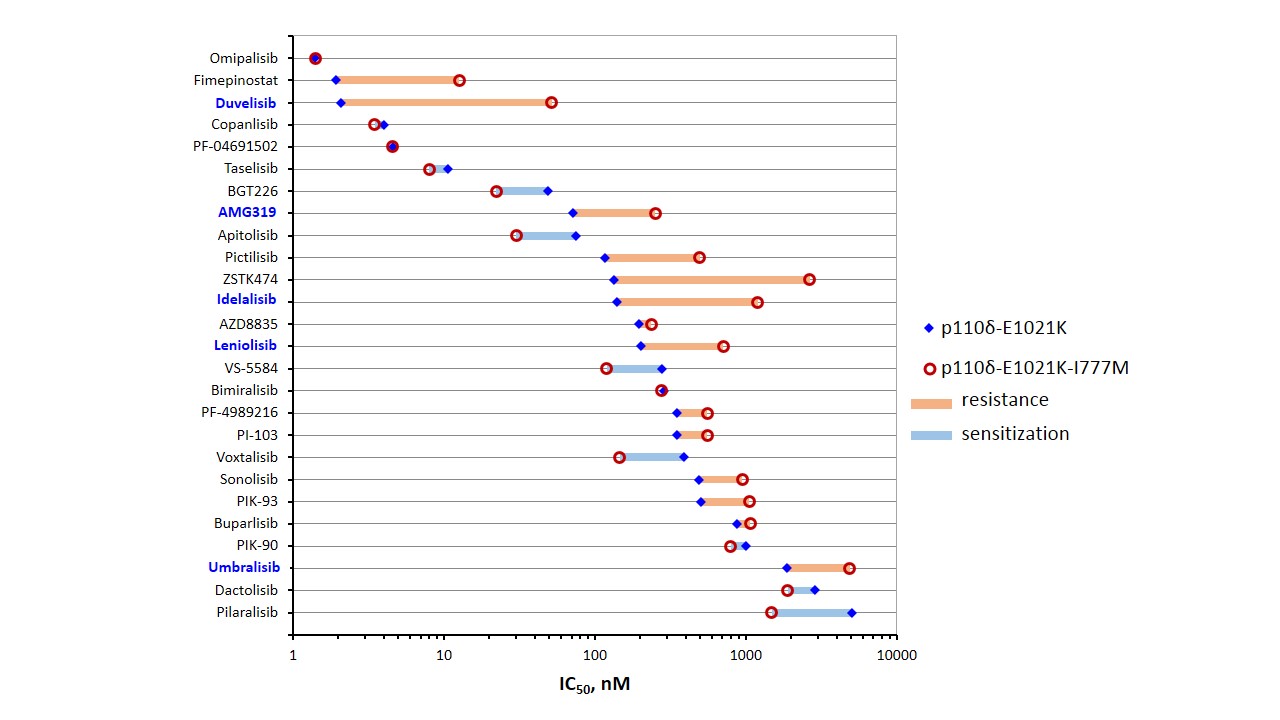 Figure 1: Cellular efficacy and mutation-mediated resistance of PI3K inhibitors. High-throughput pharmacological assessment was performed with isogenic BaF3 cells expressing mutant p110δ. PI3K inhibitors were ranked according to potency. The names of p110δ-selective substances are printed in blue.
Figure 1: Cellular efficacy and mutation-mediated resistance of PI3K inhibitors. High-throughput pharmacological assessment was performed with isogenic BaF3 cells expressing mutant p110δ. PI3K inhibitors were ranked according to potency. The names of p110δ-selective substances are printed in blue.
Among the diverse inhibitor chemotypes, inhibitor potencies as well as the effects of resistance mutations clearly distinguish multi-targeted and p110δ-selective inhibitor molecules with flat or propeller shape, respectively, according to a balance between affinity and selectivity. This simple paradigm was established, when p110δ-selective inhibitors first became available 8 and is also evident in examples of clinically approved PI3K inhibitors 9.
Structural basis of drug kinase interactions
For three example molecules we connected our pharmacological assessment with computer-guided structural investigation by molecular dynamics simulations of physical atom movements 10. This approach allows to directly follow and present the consequences of both, inhibitor interaction and genetic alterations, as shown for idelalisib binding to wild type and mutant p110δ (Video 1).
Video 1: Interaction of ATP binding pocket residues M752, W760 and I/M777 with idelalisib. In wild type p110δ (left), a specificity pocket is formed by M752 and W760 upon binding of idelalisib, whereas its formation is prevented in the same enzyme with I777M substitution (right).
According to the induced fit of propeller-shaped inhibitor molecules into the PI3K isoform p110δ discovered in co-crystal structures 11, a specificity pocket opens up between W760 and M752, when idelalisib binds. In p110δ-I777M, however, this specificity pocket to accommodate idelalisib does not form. Molecular dynamics simulations thus explain, how replacement of isoleucine 777 by the similarly sized methionine reduces idelalisib binding by affecting the flexibility of the remote M752 residue. In virtual ligand design, molecular dynamics simulations can serve to develop models that can be experimentally tested using the described cell-based systems.
A tale of two mutations
In conclusion, point mutations in p110δ turn out to be instrumental for the characterization of PI3K inhibitors: the C-terminal activating E1021K mutation by enabling cell-based functional drug assessment and the affinity pocket mutation I777M by providing insights into the molecular interactions of inhibitors with this lipid kinase isoform. As a special asset, we combine functional and structural analysis in the same study about the latter mutation.
References
(1) Owens J, 2006: High-throughput screening: Parallel lines. Nat Rev Drug Discov 5, 283.
(2) Krause G, Hassenrück F, Hallek M, 2020: Cell line-based assessment of BTK inhibitors. Br J Pharmacol 117, 2163-2165.
(3) Bishop AC, 2004: A hot spot for protein kinase inhibitor sensitivity. Chem Biol 11, 587-589.
(4) Daub H, 2010: Kinase inhibitors: Narrowing down the real targets. Nat Chem Biol 6: 249-250.
(5) Vogt, PK, 2008: Drug-resistant phosphatidyl 3-kinase: guidance for the preemptive strike. Cancer Cell 14, 107-108.
(6) Okkenhaug K, 2021: How to resist PI3Kδ inhibition: activate MAPK! Blood 138, 3-4.
(7) Vanhaesebroeck B et al., 2021: PI3K inhibitors are finally coming of age. Nat Rev Drug Discov 20, 741-769.
(8) Simon JA, 2006: Using isoform-specific inhibitors to target lipid kinases. Cell 125, 647-649.
(9) Krause G, Hassenrück F, Hallek M, 2018: Copanlisib for treatment of B-cell malignancies: the development of a PI3K inhibitor with considerable differences to idelalisib. Drug Des Dev Ther 21, 2577-2590.
(10) The Royal Swedish Academy of Sciences, 2013: The computer – your Virgil in the world of atoms. Press release, Nobel Prize in Chemistry.
(11) Workman P & van Monfort RLM, 2010: PI(3) kinases: Revealing the delta lady. Nat Chem Biol 6, 82-83.
Follow the Topic
-
Communications Biology

An open access journal from Nature Portfolio publishing high-quality research, reviews and commentary in all areas of the biological sciences, representing significant advances and bringing new biological insight to a specialized area of research.
Related Collections
With Collections, you can get published faster and increase your visibility.
Signalling Pathways of Innate Immunity
Publishing Model: Hybrid
Deadline: Feb 28, 2026
Forces in Cell Biology
Publishing Model: Open Access
Deadline: Apr 30, 2026
Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in