Artificial intelligence makes accurate quantum chemical simulations more affordable
Published in Chemistry
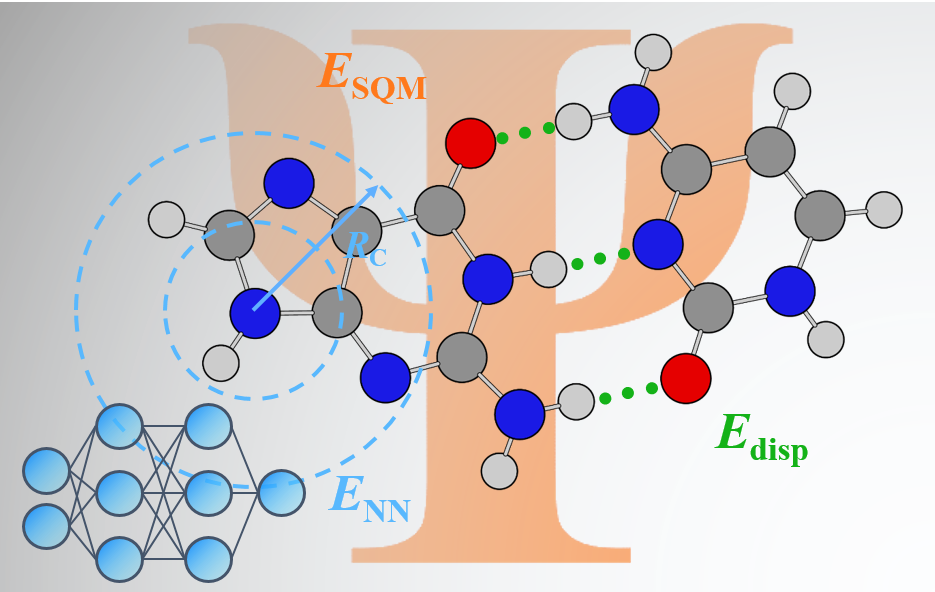

AIQM1 is a showcase of the successful combination of advances in many areas of theoretical chemistry and computational science and highlights the ongoing transition from proof-of-concept approaches based on artificial intelligence to the practical embedding of these approaches into modern quantum chemistry.
AIQM1 method is a result of a collaborative China-US effort involving two groups, one in Xiamen University and another in Carnegie Mellon University, while here I will give a personal account of what led us to the development of AIQM1. As any personal story, it starts much earlier than the start of the actual project. Before I did any research in artificial intelligence (AI, or, more specifically, in machine learning (ML) – the very tool of AI we actually used in AIQM1), in school, I was fascinated by nature and was learning about it as much as I could. I quickly realized that it is impossible to learn everything I am interested in. The more I thought about it, the more obvious was to me that the human mind is just incapable of coping with all the information, which humanity generated, and this is likely to impose a natural barrier on how we can actually make a qualitative jump in our understanding of the world. Computers, in the long run, may overcome this barrier as they are not limited by biological constraints and can have access to all digitalized information. However, can computers by themselves make a qualitative jump in our understanding of nature? This is the first question.
Later, already in the university doing my master degree, I have had to run many quantum chemical (QC) simulations. It was exciting at first to witness how a computer can predict correctly, e.g., the geometry of a water molecule, but after I run many such simulations, I realized that many of them are quite repetitive and the second question is: can computers by themselves perform all these simulations, analyze results and write papers?
I did not know the answer to these questions and did not know where to start looking for them. AI back then was still not that mature to provide answers. (Now, I would argue that we still do not know the answer to the first question, but the field is making impressive first steps to positively answer the second question.[1]) So, I made a practical decision to start with baby steps and learn first about ML as at the end of my PhD in 2013, which had nothing to do with ML, I started to come across efforts to use ML in (QC). These efforts, although at that time impressive, still showed that ML has huge problems with learning QC properties in diverse chemical space, and often, ML makes unphysical predictions. ML has an inherent problem that there is nothing in its underlying algorithm that forces it to follow the physics laws of nature; it learns from experience, and if ML did not see some particular pattern, it will fail to make correct predictions.
At the same, from my PhD work on semiempirical quantum mechanical (SQM) methods, I knew that even a rather bad SQM method may give large errors, but it has more reasonable physical behavior. For example, if you consider the dissociation curve of a diatomic molecule such as H2, we know that it has potential well, and SQM methods are capable of reproducing it, albeit with large errors. If you train ML on the right side of the minimum and do not show the region around the well, then ML will fail to predict this minimum, although it may give very accurate predictions in the region to the right of the minimum. A practical solution was to combine SQM and ML methods so that ML improves the accuracy of SQM predictions, while SQM ensures the correct physical behavior. This solution was realized in two different concepts published in 2015: one is by simply correcting SQM methods with ML (now known as Δ-learning[2]) and another one by correcting the Hamiltonian of SQM methods[3]. Both studies were proof of concept and ML has to be retrained for any new QC problem. However, already in 2013, my vision was to make a practical method, which can be used out of the box.
This vision has been shared by many researchers around the world, and in 2017 I came across the first of the universal ANI ground-state potentials, developed by the co-author of AIQM1 paper[4], Olexandr Isayev and his colleagues. ANI potentials are practical ML methods, which can be used out-of-the-box to replace costly QM methods in some simulations, and in 2019, ANI-1ccx was published[5], which used transfer learning to approach coveted coupled-cluster accuracy.
Back then I was still a post-doc swamped with other commitments such as developing a robust and accurate (within traditional SQM standards) method ODM2[6] and applying for independent positions, writing grant proposals, etc. In these applications and proposals, I was proposing to develop AI-enhanced quantum mechanical methods. Finally, I have received an offer from Xiamen University, where I was lucky to move in 2019 before the pandemic broke out. My group received enough funding from the State Key Laboratory of Physical Chemistry of Solid Surfaces and Xiamen University (and from 2021 the funding from the National Natural Science Foundation of China), so that we could afford to build our own computer cluster. At the end of the tumultuous first academic year as a newly-minted faculty, I approached Olexandr Isayev, with whom we decided to pursue together our shared vision of developing an out-of-the-box practical method by combining ideas from our previous works – ODM2, ANI, Δ-learning, and transfer learning.
 Hamiltonian, neural-network (NN) correction to the ground-state potential, and dispersion corrections.")
One crucial element of successful method development is an excellent developer and we have been lucky to find Peikun Zheng – a PhD student of Prof. Wei Wu at Xiamen University. Peikun with the help of Roman Zubatyuk from Olexandr’s group, did fantastic work within roughly one year, while also working on other research projects. One year is rather a short period of time, e.g., it took me much longer to develop ODM2, while AIQM1 tremendously improves upon previous methods compared to, e.g., a relatively modest improvement in ODM2 over the previous OM2 method. This shows just how powerful AI is and that it is already helping us to transition from proof-of-concept studies to having AI approaches as an integral part of our day-to-day QC simulations.
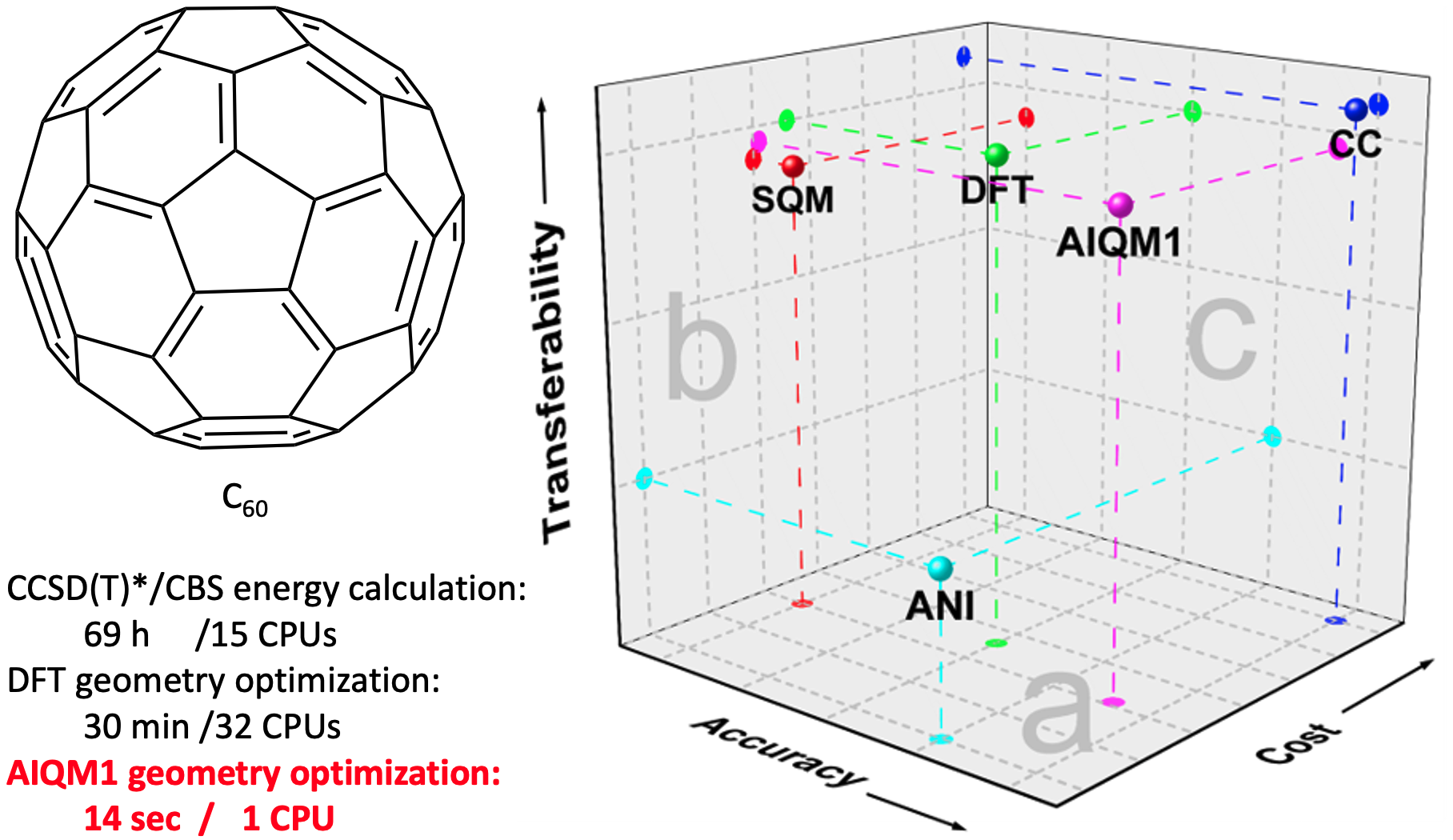
In the end, AIQM1 combined the strengths of all its components: excellent transferability and correct physical behavior of ODM2 and high accuracy of ANI. AIQM1 approaches coupled-cluster accuracy for neutral molecules in the ground state, but with the cost of SQM methods, e.g., optimization of C60 fullerene takes only 14 seconds on a single CPU, while DFT optimization takes 30 min on 32 CPUs, and single-point coupled-cluster simulations take 69 h on 15 CPUs. AIQM1 also predicts other properties, such as vertical excitation energies, with accuracy similar to popular DFT methods.
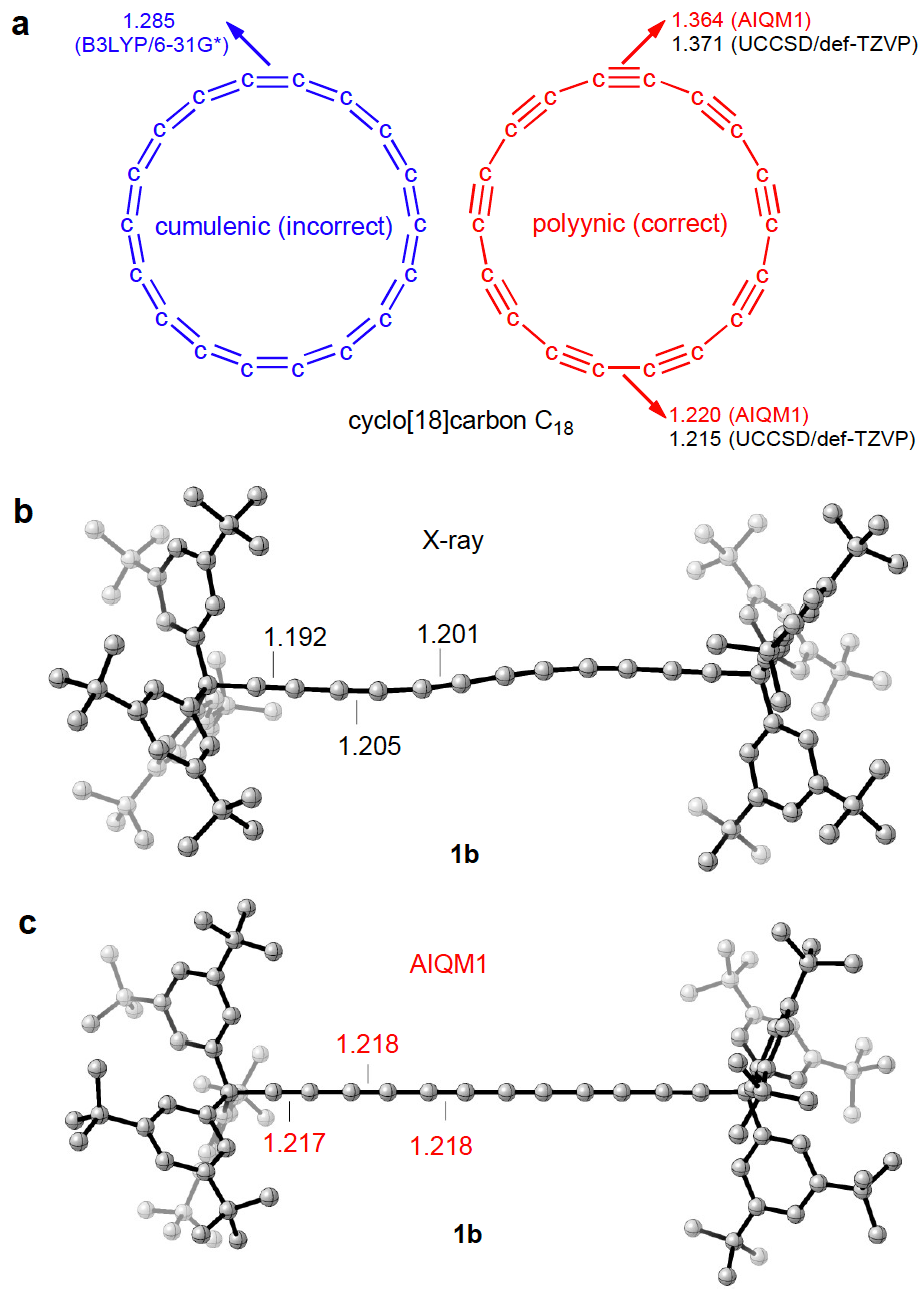
Back to the first question: “Can computers by themselves make a qualitative jump in our understanding of nature?”, I think we are still far from it, but it should not impend us from using computers to help us, humans, to make a qualitative jump in our understanding of nature. For example, while using AIQM1 to optimize geometries of a polyynic compound, we found that AIQM1 triple C≡C bond lengths are significantly longer than in X-ray structures. The computer did not make any conclusions from it, but it allowed me to make an interesting discovery that X-ray structures of such compounds are not accurate! In fact, I dug into literature and found that it is a known fact that X-ray has problems with predicting too short C≡C bond lengths. Other quantum mechanical methods affordable for performing geometry optimizations also have problems with these challenging compounds, and, e.g., popular density functional theory (DFT) approach B3LYP/6-31G* predicts incorrect cumulenic structure for cyclo[18]carbon, while AIQM1 is up to the task and predicts correct polyynic geometry. Thus, with AIQM1, we provide a tool, which allows us to make surprising discovering and rethink our understanding of nature.
P.S. I would also like to thank the reviewers of our manuscript, who made many good suggestions, which helped to make the presentation of our method clearer.
P.P.S. AIQM1 can be used with the MLatom platform interfaced with other required programs and can be obtained free of charge for non-profit uses such as academic research and education at http://MLatom.com/AIQM1. MLatom is a package for atomistic simulations with machine learning developed by our group and external contributors.
Read more in the Nature Communications paper: http://doi.org/10.1038/s41467-021-27340-2
[1] U. Raucci, A. Valentini, E. Pieri, H. Weir, S. Seritan, T. J. Martínez. Voice-controlled quantum chemistry. Nat. Comput. Sci. 2021, 1, 42–45.
[2] R. Ramakrishnan, P. O. Dral, M. Rupp, O. A. von Lilienfeld. Big Data Meets Quantum Chemistry Approximations: The Δ-Machine Learning Approach. J. Chem. Theory Comput. 2015, 11, 2087–2096.
[3] P. O. Dral, O. A. von Lilienfeld, W. Thiel. Machine Learning of Parameters for Accurate Semiempirical Quantum Chemical Calculations. J. Chem. Theory Comput. 2015, 11, 2120–2125.
[4] J. S. Smith, O. Isayev, A. E. Roitberg. ANI-1: an extensible neural network potential with DFT accuracy at force field computational cost. Chem. Sci. 2017, 8, 3192–3203.
[5] J. S. Smith, B. T. Nebgen, R. Zubatyuk, N. Lubbers, C. Devereux, K. Barros, S. Tretiak, O. Isayev, A. E. Roitberg. Approaching coupled cluster accuracy with a general-purpose neural network potential through transfer learning. Nat. Commun. 2019, 10, 2903.
[6] P. O. Dral, X. Wu, W. Thiel. Semiempirical Quantum-Chemical Methods with Orthogonalization and Dispersion Corrections. J. Chem. Theory Comput. 2019, 15, 1743–1760.
Professor in Xiamen University and Nicolaus Copernicus University in Toruń; Aitomistic, Shenzhen. Artificial intelligence for atomistic simulations.
Follow the Topic
-
Nature Communications
An open access, multidisciplinary journal dedicated to publishing high-quality research in all areas of the biological, health, physical, chemical and Earth sciences.

Related Collections
With Collections, you can get published faster and increase your visibility.
Women's Health
Publishing Model: Hybrid
Deadline: Ongoing
Tumor Microenvironment Crosstalk and Therapeutic Implications
Publishing Model: Hybrid
Deadline: Nov 02, 2026

Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in