Asymmetric alkene aminoarylations...with a Smiles:-)
Published in Chemistry
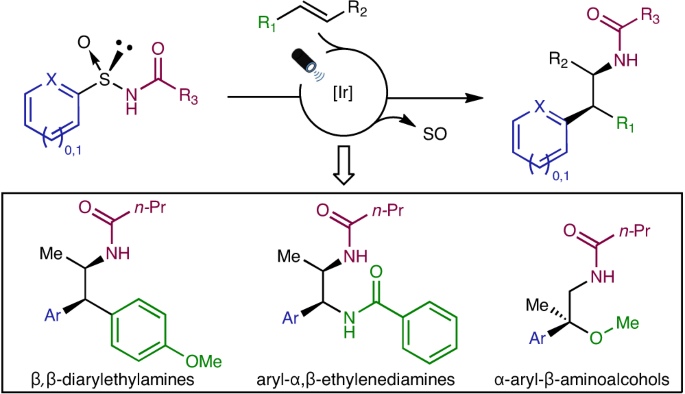
Arylethylamines are key motifs featured in a plethora of biologically active molecules. Despite their relevance, their asymmetric construction still represents a longstanding pursuit within the organic chemistry community. The use of cyclic intermediates to impart stereocontrol is an appealing strategy and thus, rearrangement reactions have emerged as prominent platforms in this context. The Smiles-Truce rearrangement has experienced a renaissance in the past few years. This transformation, orchestrated either via polar or radical mechanisms, involves the cleavage of a Csp2-X bond, followed by the formation of a novel Csp2-C bond at the ipso position via a spirocyclic intermediate (Fig. 1 left). Recently, our group described a radical Smiles-Truce rearrangement featuring N-arylsulfinyl acrylamides (Nat. Chem. 13, 327–334 (2021), Fig. 1 right). The reaction is initiated by the addition of a radical to the double bond and the N-sulfinyl group is responsible to impart absolute stereocontrol during the transposition of the aromatic ring yielding enantioenriched α-all-carbon substituted amides.
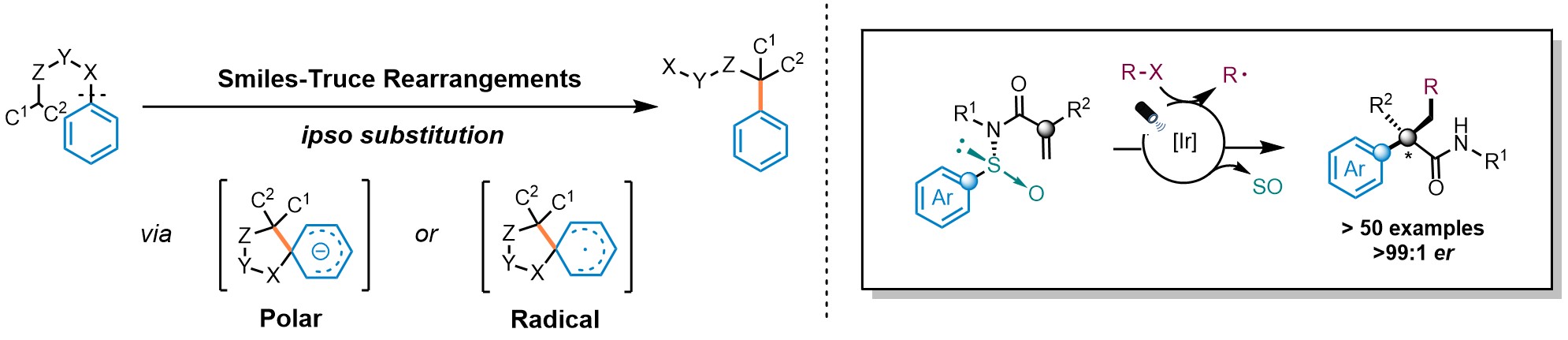
Fig. 1. Smiles-Truce rearrangement
Inspired by these results, our recent work published now in Nature Chemistry (2023, DOI: 10.1038/s41557-023-01414-8) reports a multifunctional all-in-one arylsulfinylamide reagent featuring a traceless chiral auxiliary for the asymmetric intermolecular aminoarylation of alkenes. We hypothesized that the intermolecular addition of the nitrogen atom to a 1,2-disubstituted alkene would lead to the formation of a Csp3–N bond whose absolute configuration would be controlled by the chiral sulfinyl tether. The subsequent radical Truce-Smiles rearrangement of the aryl moiety would then establish the configuration neighboring Csp3–Csp2 center. This methodology provides access to valuable β,β-diarylethylamines, aryl-α,β-ethylenediamines and a-aryl-b-aminoalcohols with excellent levels of both relative and, more importantly, absolute stereocontrol. The sulfinyl group in the reagents is eliminated in situ under mild reaction conditions. Characterization of the reaction mechanism revealed an interesting dichotomy in the initiation of the photoredox catalytic cycle wherein either electron-rich alkenes or the sulfinylamides are preferentially activated by the Ir photocatalyst at the outset of the reaction (Fig. 2).
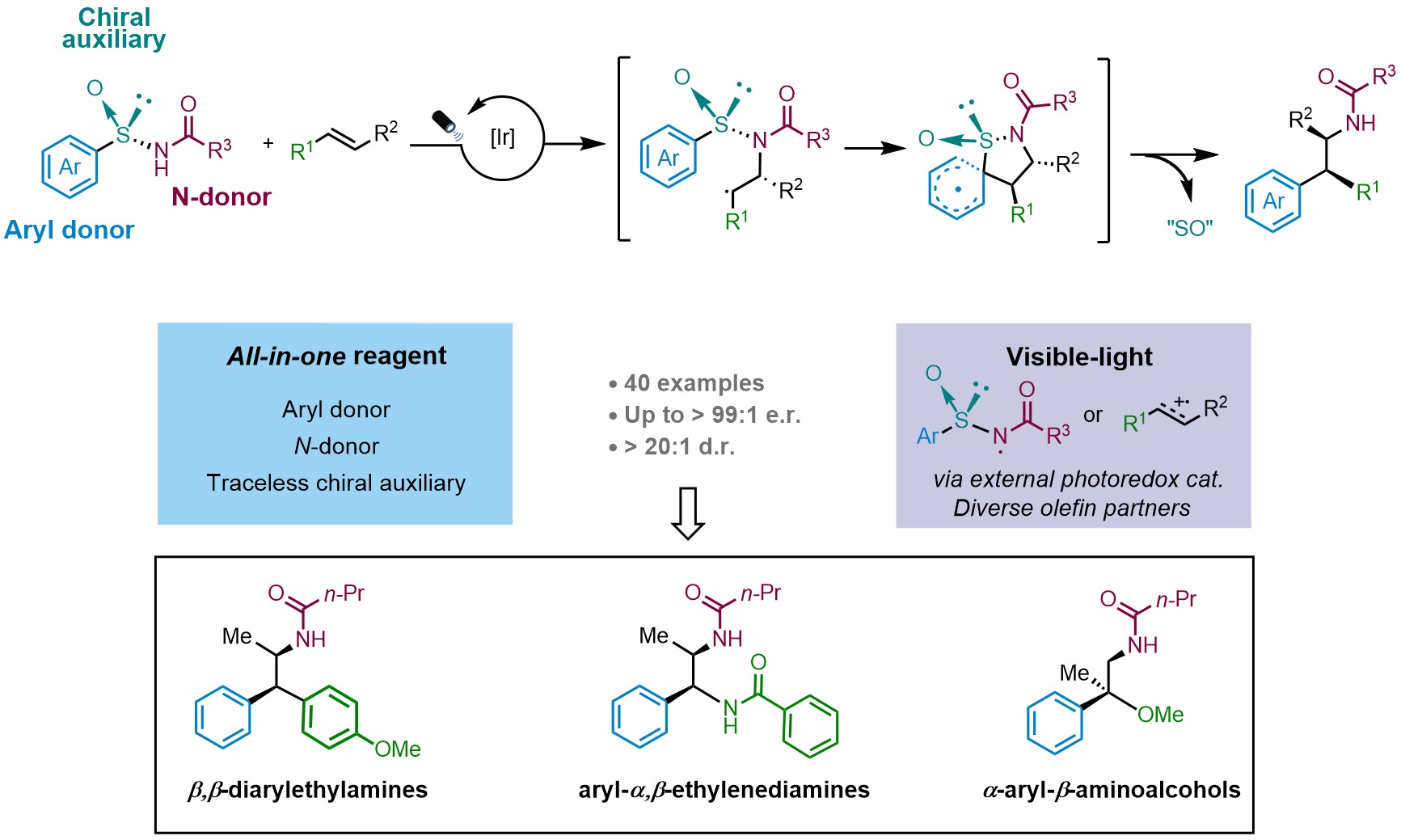
Fig. 2. Our strategy for the asymmetric intermolecular aminoarylation.
We believe that these multifunctional all-in-one reagents not only enable the assembly of relevant scaffolds populating pharmaceuticals, bioactive natural products and transition metal ligands but also open up new horizons for the efficient functionalization of diverse structural motifs.
Follow the Topic
-
Nature Chemistry
A monthly journal dedicated to publishing high-quality papers that describe the most significant and cutting-edge research in all areas of chemistry, reflecting the traditional core subjects of analytical, inorganic, organic and physical chemistry.

Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in