Atomic metal–nonmetal catalytic pair drives efficient hydrogen oxidation catalysis in fuel cells
Published in Chemistry

Hydrogen fuel cells represent promising sustainable energy conversion devices to transform chemical energy into electrical power with high efficiencies of up to 60%1. Because of heavy reliance on platinum-group metal (PGM), the cost of electrocatalysts for anodic hydrogen oxidation reaction (HOR) and cathodic oxygen reduction reaction (ORR) is projected to be the largest single component (about 40%) of the total2. In consideration of economy, ORR has witnessed enormous improvement on the atomic efficiency by pursuing single-metal-atom catalysts to replace commercial Pt/C in practical fuel cells3. Unfortunately, active single-atom electrocatalysts for HOR are still absent due to the formidable potential-dependent energy barrier for hydrogen intermediate (H*) desorption on isolated metal centers. The heavy reliance on high-loading PGM-based catalysts for HOR (Pt/C, 20~40 wt.%) largely offsets the reduced cost from the cathode side and represents a major barrier for the further progress of commercial proton exchange membrane fuel cells (PEMFCs).
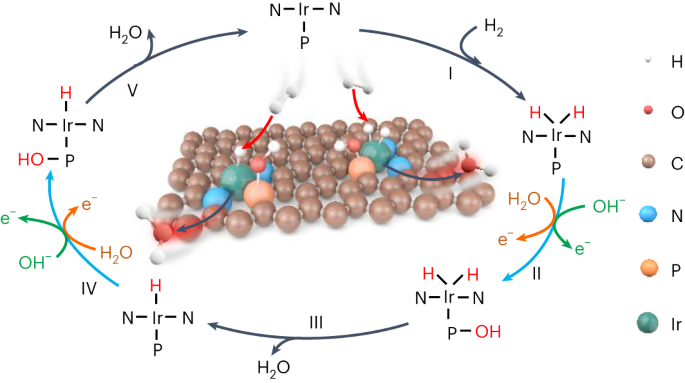
In this study, Prof. Bin Liu's group at City University of Hong Kong proposes a novel catalyst design principle of integrating paired metal–nonmetal active atoms for the first time, which introduces another reactive intermediate and induces an alternative reaction pathway to achieve an impossible catalysis process into reality on single-metal-atom catalysts. As a proof-of-concept, we construct atomically dispersed iridium and phosphorus pairs (Ir1-P1/NPG) and realize efficient and durable HOR performance, whereas the counterparts with only single-atom Ir (Ir1/NG) or P (NPG) are completely inert. A series of control experiments in conjunction with spectroscopic measurements unveil that the reactive hydroxyl (OH*) adsorbed on the more oxophilic P atomic site promotes the kinetics of H* desorption (Fig. 1). Theoretical calculations further demonstrate that the formation of H2O from OH* and H* on adjacent P and Ir atoms is indeed a thermodynamic redox process, which can be activated by ambient temperature without additional requirement of applied overpotential.

Structural identification of atomic Ir1-P1 pairs
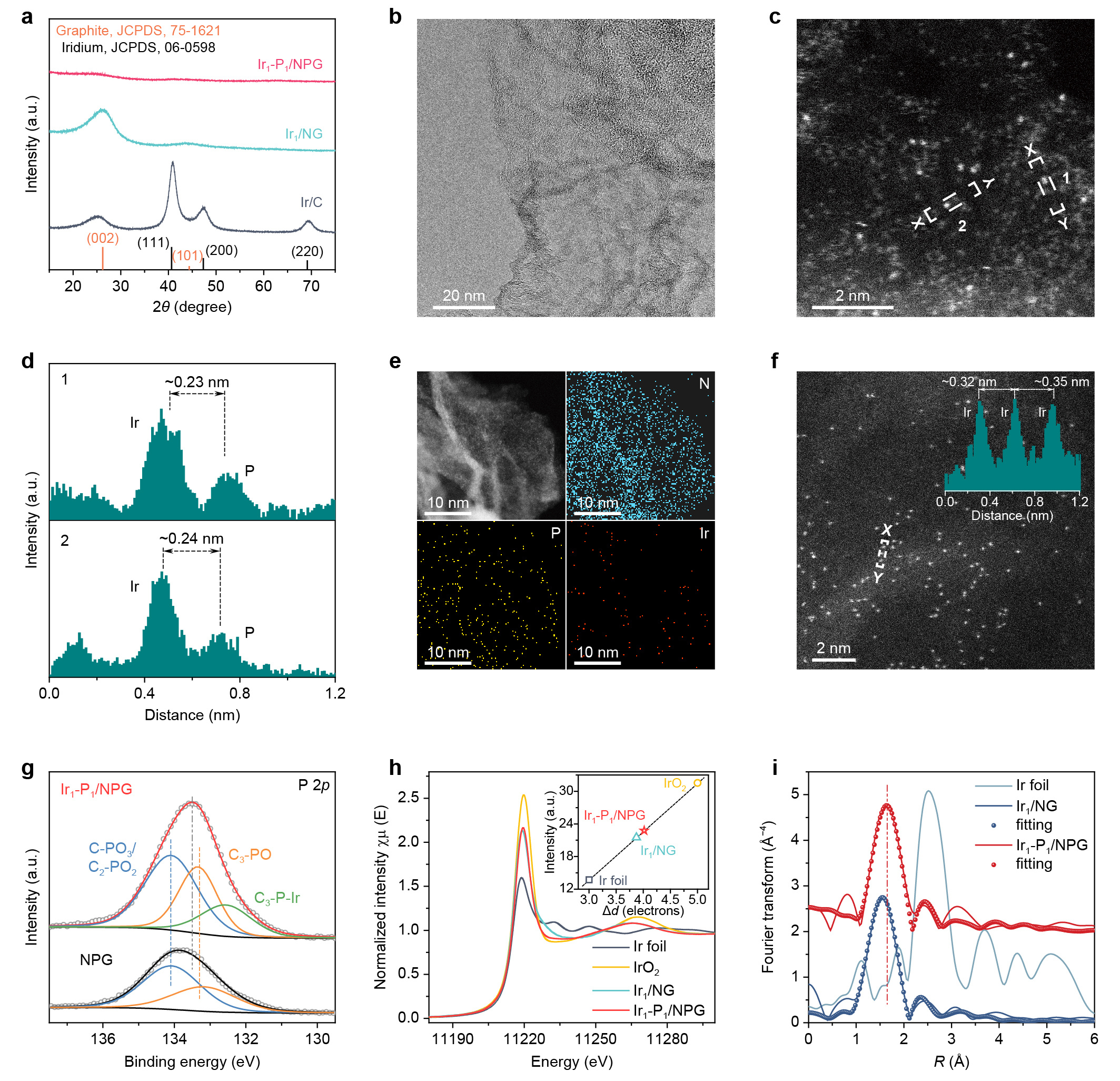
To provide sufficient evidence to justify the atomic dispersion and interaction between Ir and P, we firstly performed XRD, survey XPS and EDS measurements to identify the elemental compositions of our catalysts. Then, through comparing HAADF-STEM images (including their element-specific electron scattering cross-sections) and quantitative STEM simulations of Ir1/NG and Ir1-P1/NPG, atomically dispersed pairs of Ir and P could be identified in Ir1-P1/NPG. Furthermore, the coordination interaction between paired Ir and P atoms in Ir1-P1/NPG was verified by P 2p XPS, EXAFS fitting as well as PDOS analysis (Fig. 2).
Catalytic performance of Ir1-P1 CPs in HOR and PEMFC
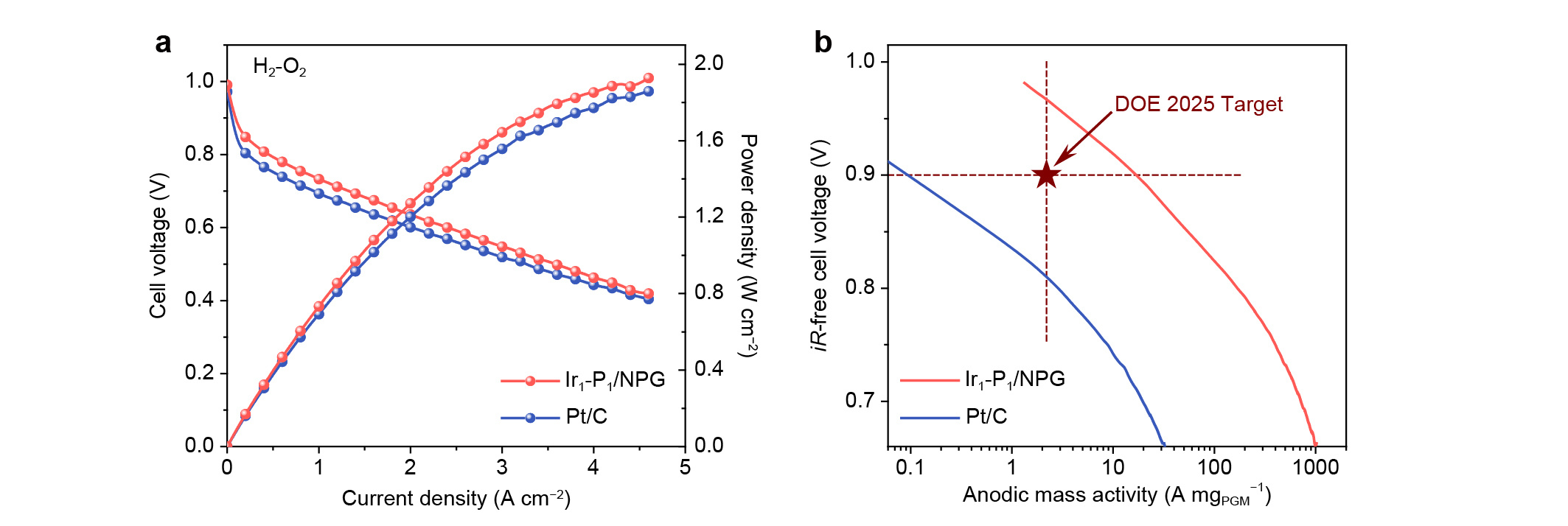
The HOR performance of Ir1-P1/NPG and Ir1/NG were evaluated in both H2-saturated aqueous 0.1 M HClO4 and 0.1 M KOH on a rotating disk electrode (RDE). Typical HOR polarization curves with constant diffusion-limited currents are observed for Ir1-P1/NPG, whereas Ir1/NG exhibits negligible HOR activities. Through fitting the kinetic current densities in alkaline electrolyte, we find that the exchange current density of Ir1-P1/NPG is not only about three times and one order of magnitude greater than that for Pt/C and Ir/C with the same PGM loading amount, but also stands at the top level among the state-of-the-art HOR catalysts that have been reported so far. More importantly, the performance of our Ir1-P1/NPG catalyst as the anode for PEMFC is unprecedented (Fig. 3): the anodic mass activity at 0.9 ViR-free reaches 17.11 A mgPGM−1, which is nearly eight times higher than the U.S. Department of Energy (DOE) 2025 target.
Mechanism understanding of Ir1-P1 CPs
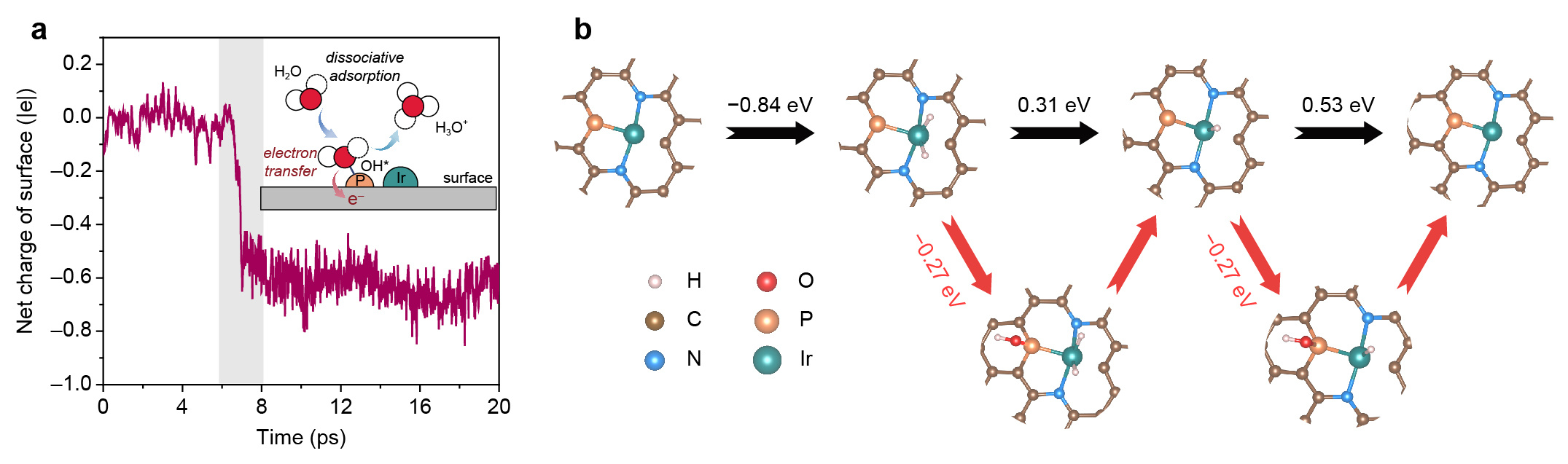
By selectively poisoning Ir and P sites, the HOR activities of Ir1-P1/NPG were significantly suppressed, indicating the indispensable role of both Ir and P for HOR. Besides the hydrogen adsorption energy determined by Ir atoms, it was interesting to note that there were hydroxyl species (OH*) adsorbed on P sites during HOR, observed in quasi in-situ XPS spectra. Mulliken charge analysis also showed that such adsorption of OH* on P was indeed a proton-coupled electron transfer step evidenced by an accumulation of −0.7|e| on catalyst's surface (Fig. 4a). Thus, in the OH*-participated HOR route, the explicit electron transfer step occurs on the oxidative adsorption of OH* rather than the desorption of H*. To determine whether OH* adsorbed on P sites could participate in electrochemical reaction, we performed CO stripping test with and without quenching P sites, revealing that it is the OH* adsorbed on P sites that oxidatively reacts with CO. As a result, it is plausible to propose a bifunctional HOR mechanism over integrative Ir1-P1 CPs, that is, HOR occurs through dissociative adsorption of H2 on Ir sites followed by combination with the reactive OH* adsorbed on the adjacent P sites (Fig. 4b).
Bigger picture of integrative CPs concept
In this work, Dr. Qilun Wang, Prof. Hong Bin Yang, Prof. Bin Liu and co-authors proposed the integrative metal-nonmetal catalytic pairs (CPs) concept for the first time, and demonstrated their fascinating catalytic activity and mechanism. The same concept can be extended to other metal-nonmetal, metal-metal and even nonmetal-nonmetal CPs to drive chemical reactions involving multiple intermediates based on distinct demands of active sites. It is hoped that this story will be of immediate interest for broad readers in both basic and applied communities.
References
- Stamenkovic, V. R., Strmcnik, D., Lopes, P. P. & Markovic N. M. Energy and fuels from electrochemical interfaces. Nat. Mater. 16, 57–69 (2017).
- Debe, M. K. Electrocatalyst approaches and challenges for automotive fuel cells. Nature 486, 43–51 (2012).
- Mehmood, A. et al. High loading of single atomic iron sites in Fe–NC oxygen reduction catalysts for proton exchange membrane fuel cells. Nat. Catal. 5, 311–323 (2022).
Follow the Topic
-
Nature Catalysis
This journal brings together researchers from across all chemistry and related fields, publishing work on homogeneous catalysis, heterogeneous catalysis, and biocatalysts, incorporating both fundamental and applied studies.




Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in