Autocatalysis Driven by Solvent-Free Supramolecular Polymerization
Published in Electrical & Electronic Engineering


The idea of an entity that can continuously produce replicas of itself has captured the imagination of many thinkers, from von Neumann to Vonnegut. Essentially, this process can be described as autocatalysis. An autocatalytic reaction is one in which the product acts as a catalyst for its own formation. Autocatalysis has been implicated in the emergence of life and it is intrinsic to many biological processes, such as the self-replication of biomolecules.
Scientists have long tried, using diverse strategies, to achieve autocatalysis in chemical transformations. Consider for example (Fig. 1), a reaction product designed to serve as a template (T) that can reversibly preorganize reactants A and B in the form of a ternary complex [A•T•B]. This facilitates the reaction between A and B to produce T•T. However, if T•T does not serve as a template, the expected autocatalytic behaviour would not emerge unless T•T dissociates into monomeric T. This is so called "product inhibition". Such decrease in active template concentration due to product inhibition and/or template aggregation is an essential problem in nonbiological organic autocatalysis and usually disturbs the expected sigmoidal time-course profile (Fig. 1). On the contrary, inorganic redox chemistry may involve inhibition-free autocatalysis, with the famous Belousov-Zhabotinsky chemical oscillator being a notable example. We also know that inorganic nanocrystals form in an inhibition-free autocatalytic manner, where precursor metal ions in aqueous media preferentially adsorb onto the surface of seed nanocrystals and are electronically reduced to become a part of the growing nanocrystals.
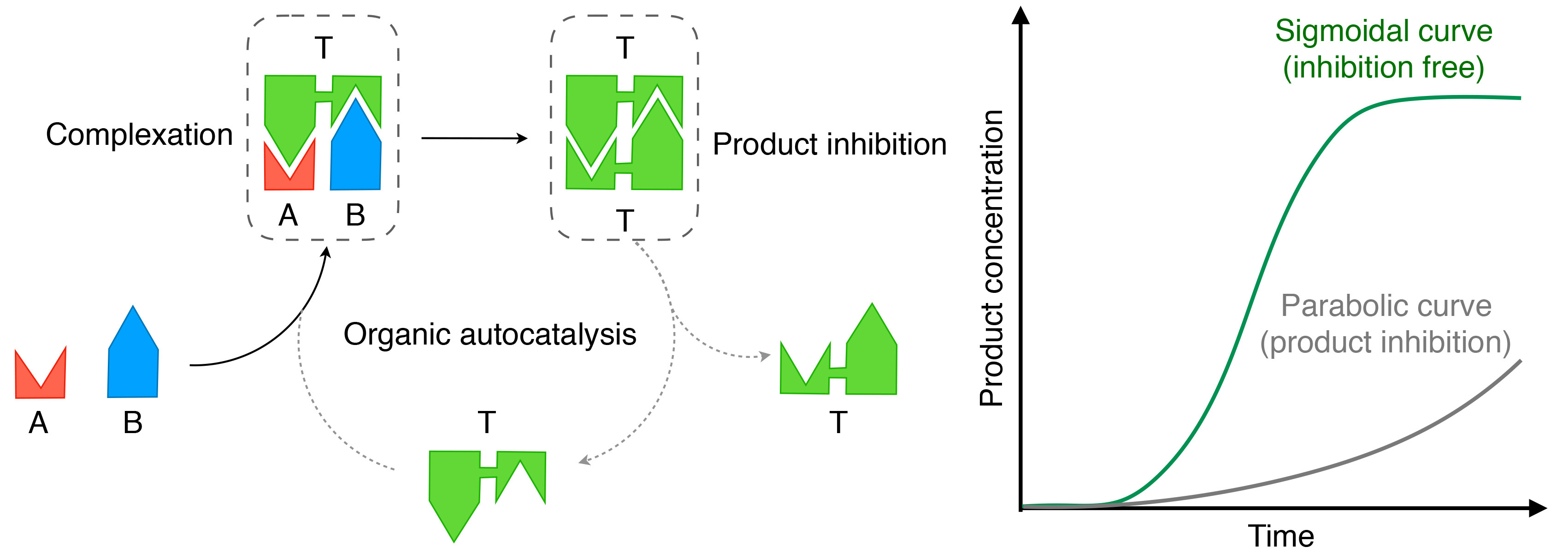
How can inhibition-free organic autocatalysis be achieved? In 2010, Otto and coworkers provided a possible solution. They found that in the reversible oxidative cyclization of a dithiol having a β-sheet-forming peptide spacer, macrocyclic products with certain ring sizes are selectively produced with a typical sigmoidal time-course profile if a shear force is continuously applied to the reaction mixture (J. M. A. Carnall, et al., Science, 327, 1502 (2010)). In their case, the macrocycles selectively produced serve to template the macrocyclization. However, they tend to stack into nanofibres in the reaction medium, and such nanofibres also tend to combine together end to end. Consequently, the concentration of active templates decreases, thereby hampering the autocatalytic process. However, if a shear force is continuously applied, nanofibres can be cut into numerous short pieces with active ends for templating the reaction. In this manner, Otto and coworkers successfully avoided the product-inhibition effect. Because the oxidative cyclization they used is intrinsically reversible, they have also suggested the possibility that the observed selectivity might be partly due to an equilibrium shift caused by the removal of preferred products as nanofibers form.
In our recent work, we provide a clearer solution. The solvent-free autocatalytic supramolecular polymerization (SF-ASP; Fig. 2) reported here provides an inhibition-free template-assisted catalytic organic transformation that takes great advantage of the fact that the product (template) undergoes a termination-free nucleation-elongation assembly (living supramolecular polymerization) under solvent-free conditions. The monomers for SF-ASP are phthalocyanine (PC) derivatives, which are produced via reductive cyclotetramerization of their phthalonitrile (PN) precursors that adopt a fan shape with hydrogen-bonding (H-bonding) amide groups. In SF-ASP, any PC monomer produced nucleates and initiates H-bonding-mediated supramolecular polymerization to give one-dimensional single-crystalline fibres, which can preorganize PN molecules via an H-bonding interaction at the cross-sectional fibre end and efficiently template their reductive cyclotetramerization to give PC in very high yield (>80%). With metal oleates, SF-ASP produces single-crystalline fibres of metallophthalocyanines, again in exceptionally high yields, which grow in both directions, without terminal coupling, until PN precursors are completely consumed. By taking advantage of the living nature of this polymerization, multistep SF-ASP with and without metal oleates allows for the precision synthesis of multi-block supramolecular copolymers.
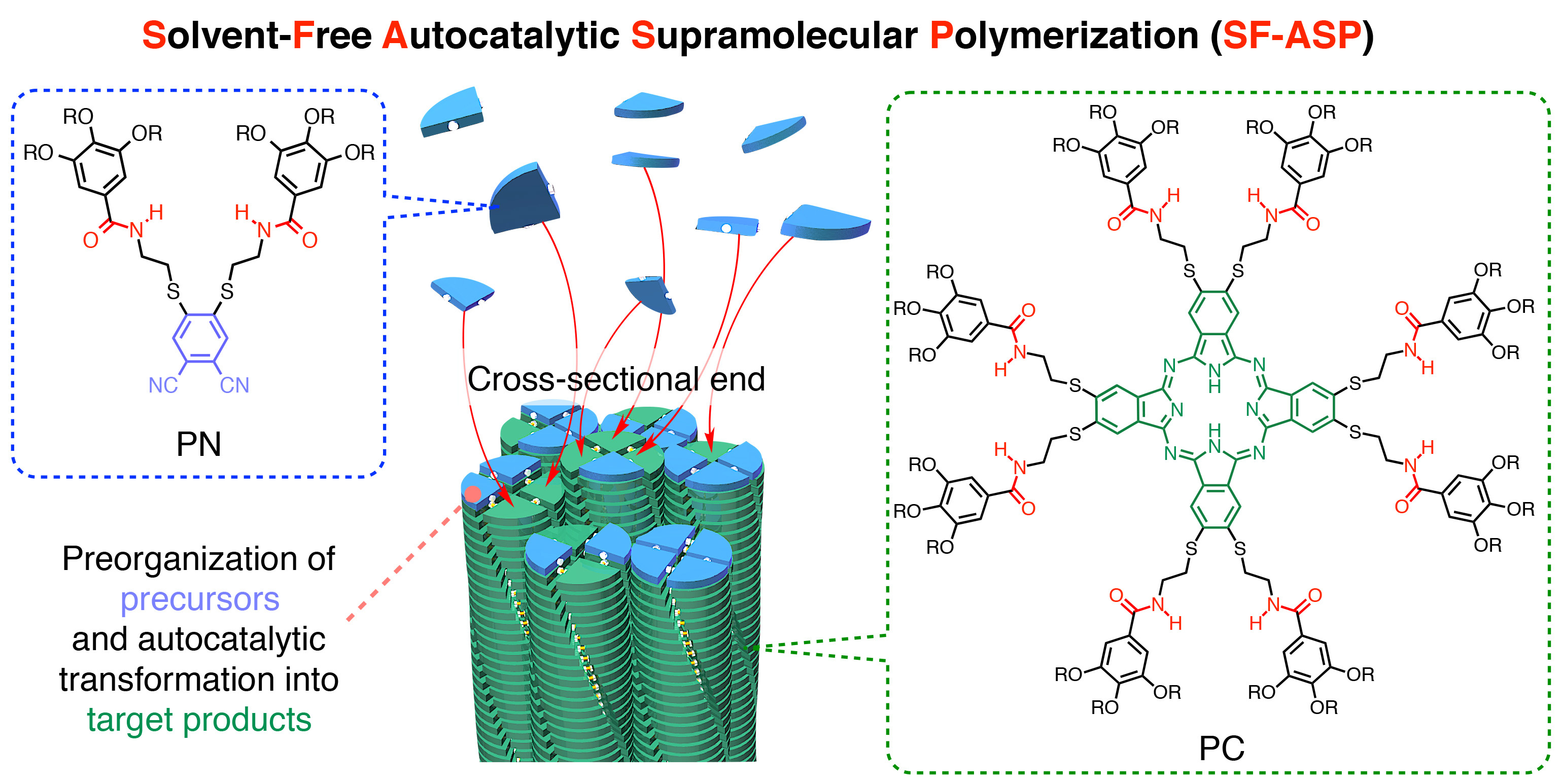
Why does SF-ASP proceed in such perfect autocatalytic manner? As illustrated in Fig. 2, the cross-sectional fibre ends preorganize the PN molecules via H-bonding interactions, thereby facilitating their efficient reductive cyclotetramerization into PC. Then, a new set of PN molecules can likewise be preorganized on the newly formed cross-sectional fibre end. Repetition of this sequence of elementary steps results in fibre elongation. We monitored the above process (Fig. 3) and found that fibres emerged 3–4 hours after heating, and then continuously elongated and increased in their number and thickness over a further period of 4 hours. Thereafter, the formation of new crystalline fibres subsided, but the formed fibres continued growing thicker, increasing the total cross-sectional area, and thereby further promoting the chemical transformation of PN into PC. In other words, PC is continuously produced, without product inhibition, at the cross-sectional fibre ends until PN is completely consumed. Equally important, end-to-end coupling of the crystalline fibres, which would result in a decrease in the total cross-sectional area for templating the reaction, barely occurred in SF-ASP, likely due to their very sluggish diffusion in the hot melt of PN under solvent-free conditions.

The basic principle of SF-ASP was found serendipitously, stemming from a mistake made during our study on the ferroelectric nature of H-bonding PN derivatives. In 2012, our group designed a columnar liquid crystal adopting a core–shell architecture showing a ferroelectric response (D. Miyajima, et al., Science, 336, 209 (2012)). This particular structure consisting of four PN molecules accommodates an array of polar cyano groups confined by a H-bonded column. Under an applied electric field, both columns and core cyano groups align unidirectionally, thereby developing an extremely large macroscopic remnant polarization. Mistakenly, we forgot to stop heating a PN derivative, keeping it at 160 °C overnight, and found, very surprisingly, that green-coloured thin fibres formed and elongated on the hot stage. Seeing the growth of such beautifully coloured fibres attracted our big interest and motivated us to study the details of what happened. By matrix-assisted laser desorption ionization time-of-flight mass spectrometry, we eventually found that the fibres obtained were composed of PC. Building on this serendipitous discovery, we report SF-ASP, which allows for precision macromolecular engineering using in situ produced monomers under solvent-free conditions. We believe that SF-ASP could become one of the ideal forms of polymer manufacturing for the sustainable future.
For more information, please refer to our recent publication in Nature Materials, "Solvent-free autocatalytic supramolecular polymerization".
Dr. Zhen CHEN is currently Assistant Professor at Tsinghua Shenzhen International Graduate School (Tsinghua SIGS), Tsinghua University. He received his Ph.D degree from the University of Tokyo, in 2016 under the supervision of Prof. Takuzo Aida, and then worked as a postdoctoral fellow in Prof. Vivian Wing-Wah Yam’s group at the University of Hong Kong during 2017–2021. Dr. Chen’s research interests mainly focus on the functional organic polymers and supramolecular self-assembled soft materials. He has published papers, including in Nature Materials, Nature Communications, PNAS, J. Am. Chem. Soc., Angew. Chem. Int. Ed. etc., and applied patents. He previously got Japan Government MEXT Scholarship.
Follow the Topic
-
Nature Materials

A monthly multi-disciplinary journal that brings together cutting-edge research across the entire spectrum of materials science and engineering, including applied and fundamental aspects of the synthesis/processing, structure/composition, properties and performance of materials.




Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in