Bacterial membrane targeting to improve antibiotic activity
Published in Microbiology

The paper in Nature Communications is here: http://go.nature.com/2DivGwi
I became interested in antibiotic research during my travels in South East Asia, India, Nepal, Pakistan and the Middle East in my youth, where sadly I saw many people die from ‘treatable’ infections, including a young woman in the Nusa Tengarra islands of Indonesia who died soon after childbirth in a village with no clean water, electricity or rudimentary health care.

I would then always travel with a sizable ‘field-kit’ of antibiotics, bandages, sutures etc. and give them to local nurses or doctors where I could. Whilst we are focused on the overuse of antibiotics driving resistant, it is important to remember that many disadvantaged people in the world don’t have access to any antibiotics; one of the reasons why neonatal and postpartum death rates are so high outside of the OECD countries.

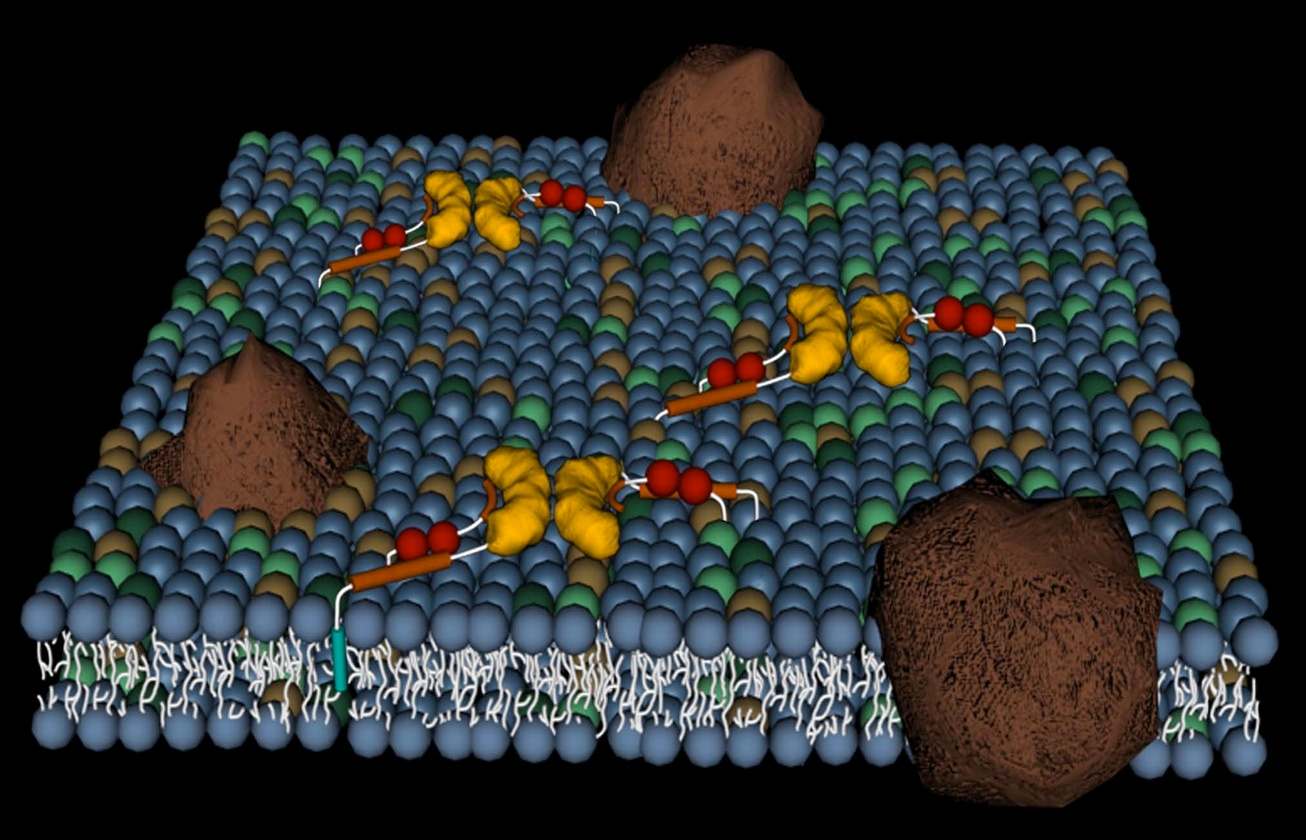
After travelling overland from Australia to Istanbul, I arrive in Cambridge UK as a postdoc in Prof. Dudley William’s group at the University working on molecular recognition. I became interested in membranes and how proteins and drugs interacted with them. Sitting in one of the college gardens for a lunch break, I wondered what would happen if we took the membrane-selective components of peptides and proteins and chemically ligated them to an out-of-patent drug, that was already given intravenously, but that had problems with toxicity or resistance. The idea was to increase the effective concentration of the drug chemotype at the membrane of the target cell to minimise side effects and maximise biology action. As I was already studying vancomycin and the glycopeptide antibiotics and mechanisms of resistance to them, this quite old, but still powerful drug seemed like a good starting point.
The vancapticins are a new class of glycopeptide antibiotics, modified versions of the archetypal glycopeptide vancomycin which has been modified to selectively target bacterial membranes. Unlike other classes of antibiotics containing multiple members, there are very few glycopeptides used in the clinic. Vancomycin and teicoplanin were first used in the 1950s and 1980s, respectively, with three new semisynthetic derivatives recently approved: telavancin in 2009 and dalbavancin and oritavancin in 2014. These three new analogs had complex and extended development timelines, passing through multiple owners and delays before eventually reaching the market.
The University filed IP on my idea and was licensed to a Cambridge-based startup, Adprotech Ltd, a company focused primarily on complement inhibition to treat inflammation. In 2004, Adprotech was acquired by Inflazyme Pharmaceuticals Ltd, based in Vancouver B.C. As Inflazyme was also primarily focused on novel anti-inflammatory therapies, the antibacterial aspect of the membrane targeting technology was largely neglected.
In 2009 I was awarded an NHMRC Australian Fellowship to return to Australia at the Institute for Molecular Bioscience at The University of Queensland, based on a proposal to establish a research group focused on fighting the emerging threat of antimicrobial resistance. As part of this proposal, I negotiated to retrieve the intellectual property rights to the vancapticin program from Inflazyme. I also was successful at obtaining a local NHMRC project grant to kick start the research to improve our original ideas, predominantly by converting the labile disulfide attachment employed for proof of concept studies, to a more stable and drug-like carbon-based linkage.
Data obtained from these studies provided enough evidence to support an ultimately successful application to the Wellcome Trust Seeding Drug Discovery program. This enabled a significant increase in the resources allocated to the program, including the hiring of additional senior researchers with extensive industry drug development expertise, including Dr. Mark Blaskovich.
Over the course of three years we made substantial progress in improving the vancapticins, creating over 300 analogues that varied the membrane selective and linker components, thereby identifying promising lead compounds potentially suitable for progression into formal preclinical development. To do this we had to build a competitive team covering microbiology, ADMET and the requisite pre-clinical models of infection. A powerful byproduct of this research program is CO-ADD, the Community for Open Antimicrobial Drug Discovery.
We leveraged the antibiotic expertise and facilities enabled by the initial Wellcome Trust funding to establish an open access crowd-sourcing approach to discovering new antibiotics, offering free antimicrobial screening to academic chemists around the world. Hopefully, the vancapticins, CO-ADD and other programs like them can eventually help the people in need that first inspired my journey.

Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in