
If mutating multiple sites of a protein is like spinning the slot-machine, how do we know the chance of hitting a jackpot without a “paytable”?
When we rationally design our favorite protein, like Cas9 used in the CRISPR system, based on its structural information, we may end up choosing multiple sites of the protein sequence for engineering. Multiple amino acid changes may be required together to give a significant improvement and individual change may be neutral or even deleterious to the protein function. The number of combinations increases dramatically with every additional amino acid to be modified. If each mutagenized part resembles an individual reel slot in a slot machine while spinning the reels is like substituting one part into another or switching a residue into other amino acids, then referring to the “paytable” as a guide to do multiple site-directed mutagenesis will potentially lead us to the winning combination of mutations for protein engineering.
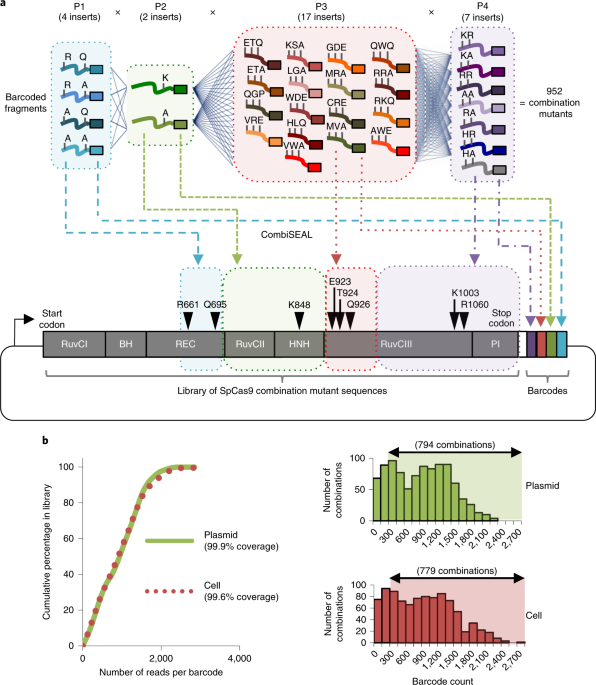
CombiSEAL can construct this “paytable” by tracking the relative frequencies of every barcoded protein variants in a library during the selection process using the NGS quantitative readout.
Often after a selection process, we have no clue how each of the protein variants is enriched or depleted. This limits our ability to determine if the selection pressure is optimal and whether we have selected the best variant from the pool. We cannot tell if the selection is successful until the positive clones are uncovered and compared against the wild-type protein. However, the throughput of isolating and sequencing the positively selected clones is very limited in guiding us through the selection process. To address the above concerns, we have designed the CombiSEAL screening platform for tagging the protein-coding sequences so that we can then leverage on next generation sequencing (NGS) for a quantitative readout to determine the abundance of each protein variant relative to the wild type during the selection. An additional advantage is that the same barcoded library can be selected again under different conditions or by using other reporter systems for selecting alternative desirable phenotypes. An analogy of this new screening platform CombiSEAL is like spinning the slot-machine many times to screen through all possible combinations of pre-selected mutations at intended positions. From there, we work backwards to construct a detailed paytable by matching each combination to its payout. This paytable also helps make systematic comparison among variants and distinguish the major contributing mutations for improving protein performance.
Why choose CombiSEAL? And, how does it work?
If the mutating sites are far from each other and synthesizing a very long fragment for cloning a library of protein variants in a pool is not feasible, combinatorial Golden-Gate-based strategies might help. However, it also requires long-read sequencing to recover the identity of each variant. Deploying long-read sequencing for tracking mutants with high sequence similarity is hard due to its high cost and error-rate. To overcome this constraint, CombiSEAL was developed as a platform to modularize protein into multiple segments for generating mutated variants with barcodes by seamlessly ligating the mutagenized parts and concatenating their corresponding barcodes locating on one end. The short-read sequencing output of these barcodes can rapidly track a massive number of protein variants in parallel and thereby allow us to construct a “paytable” for the sequence-activity relationship of a protein. Using this platform, we discovered high-fidelity Cas9 variants, Opti-SpCas9 and OptiHF-SpCas9, which are compatible with gRNAs containing an additional 5’ guanine required for its efficient expression under the widely-used U6 promoter.
With this new approach for assembling and tagging a library of protein variants, we are able to evaluate the performance for each variant under different conditions in an unprecedented throughput. In the future, with more CombiSEAL protein libraries available for analyses, we may find the rule for identifying a “loose slot machine” with better payouts.
Read the paper here: https://www.nature.com/articles/s41592-019-0473-0
Written by Gigi Choi
Image by Michael Yip
Gigi would like to acknowledge Michael Yip for the image and the slot machine analogy; Alan Wong, Yukkei Wan, and other AW lab members for the suggestions and comments for writing this blog.
Follow the Topic
-
Nature Methods
This journal is a forum for the publication of novel methods and significant improvements to tried-and-tested basic research techniques in the life sciences.

Related Collections
With Collections, you can get published faster and increase your visibility.
Methods development in Cryo-ET and in situ structural determination
Publishing Model: Hybrid
Deadline: Oct 30, 2026

Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in