Behind the paper: Chemical generation of checkpoint inhibitory T cell engagers for the treatment of cancer
Published in Chemistry

Over the past few decades, the advent of immunotherapy has revolutionized how cancer is treated.1 In particular, antibody-based therapies have become a mainstay of the oncologist’s toolbox. The premier example of these strategies is checkpoint inhibition, where an antibody targets an immune checkpoint protein – a cell surface receptor whose function is to downregulate the immune response, essentially acting as an “off” switch. Cancer cells have evolved to exploit this and thus overexpress checkpoint inhibitory receptors to evade destruction by the immune response. However, antibodies can bind to these receptors, blocking their function and thereby restoring the capacity of the immune system to fight the cancer. Another example of using antibodies (or their fragments) to modulate the immune response is that of bispecific T cell engagers. Here a so-called bispecific antibody is generated, which (as the name suggests) is capable of binding to two targets at the same time. These targets are usually a cancer antigen (for targeting) and a T lymphocyte. The T cell is thereby held close to the tumour cell, is activated (through binding of its activatory CD3 receptor), and subsequently kills the malignant cell. Examples of both strategies have obtained FDA approval.1,2
Bispecific antibodies (bsAbs) are usually generated with protein engineering – i.e., a recombinant amino-acid sequence is encoded in a suitable expression vector producing a “Frankenstein’s antibody”. These methods are now widely applied in industry. However, in recent years, advances in protein bioconjugation (the modification of proteins with small molecules) and bioorthogonal click chemistry are bringing chemistry-based methods into parity with recombinant strategies.3 Chemical strategies offer advantages in some areas such as increased modularity, speed of assembly, and the inherent ability to further modify the bsAb (i.e., with additional proteins or small molecules). We have previously developed such a method where two antibody antigen-binding fragments (Fabs), derived from native monoclonal antibodies, are modified via disulfide-rebridging to incorporate complementary click handles (tetrazine and strained alkyne, respectively). These functionalized Fabs are then clicked together via an inverse electron-demand Diels-Alder cycloaddition to form a Fab-Fab bsAb.4
In this work, we envisaged significantly expanding the previous method by attaching an additional protein to the bsAb core. Such functionalized three-protein conjugates have not been generated with chemistry before (with the only other example of three separate proteins clicked together fundamentally limited to an IgG-like scaffold).5 Furthermore, inspired by previous reports where protein engineering was used to generate a so-called checkpoint inhibitory T cell engager (CiTE) construct where a T cell engager core was combined with inhibition of the PD-1/PD-L1 pathway,6 we set out to provide a method for the chemical generation of functionalized CiTEs.
We have successfully generated two CiTE molecules. Both comprise a T cell engager core, with an anti-CD3 (binds to T cells) and an anti-HER2 (binds to cancer cells) Fab clicked together. CiTE 24 then has a sialidase enzyme attached, which has been shown to remove immunosuppressive sialic acid glycans from both immune and cancer cells, increasing immune activation.7 CiTE 27 instead contains a 3rd Fab, which binds PD-1, disrupting the aforementioned PD-1/PD-L1 pathway. Additionally, both CiTEs contain a biotin small molecule to aid imaging. This biotin moiety could be substituted to any other clickable small molecule (dye, drug, radiotracer, masking species, etc.) highlighting the modularity of the method.
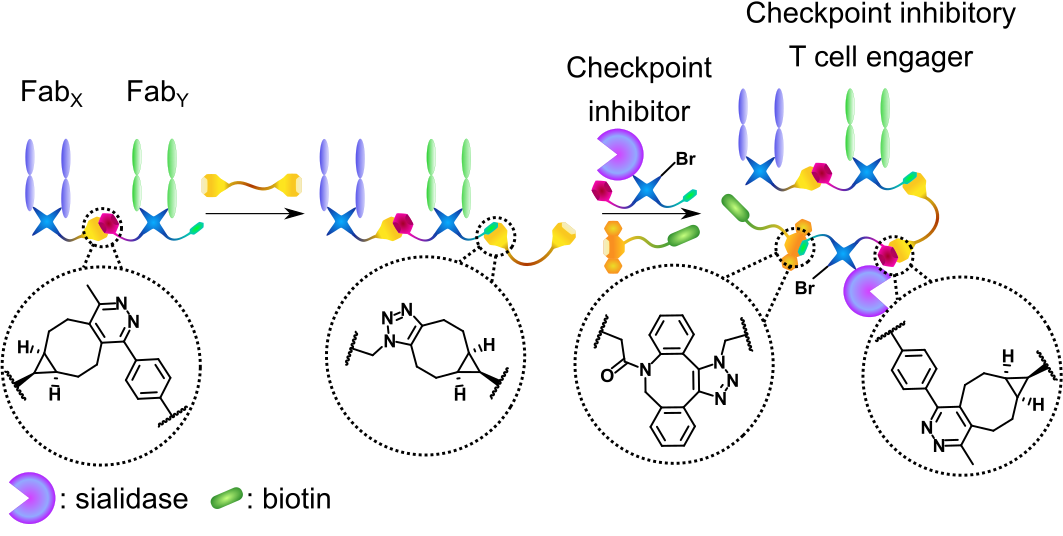
Figure 1 | Chemical generation of a checkpoint inhibitory T cell engager.
The antigen-binding fragments of antibodies (Fabs) or a sialidase enzyme are decorated with bioorthogonal click handles and clicked together. A biotin small molecule is added as a 4th component.
These CiTE species were then compared to the corresponding anti-CD3/anti-HER2 bispecific T cell engager (BiTE, also generated via similar chemistry), to assess whether the biological functionality of the component proteins was preserved. Gratifyingly, all the components showed retention of biological activity, with the caveat, that whichever Fab was in the middle of the species (anti-CD3 for CiTE 24 and anti-HER2 for CiTE 27) showed reduced binding to its target receptor, presumably due to steric hindrance. This could provide a way of modulating the biodistribution of the conjugate – as for instance a lower CD3-binding would be beneficial to limit off-target accumulation and immune activation.
The CiTEs were then compared to the BiTE in a co-culture assay between HER2+ MDA-MB231 cells and human T cells in vitro. It was found that both CiTEs showed significantly enhanced cytotoxicity over the BiTE, with the sialidase enzyme-containing CiTE 24 being especially potent.
In summary, we have developed a modular chemical method for the generation of functionalized three-protein conjugates. Furthermore, we generated two functional CiTE molecules. The protein components of the CiTEs retained their biological function and showed enhanced T cell-mediated cytotoxicity in vitro compared to the BiTE core. The sialidase-containing CiTE 24 has shown particular promise with significantly enhanced potency, suggesting it as a candidate for further investigation as an immunotherapeutic modality.
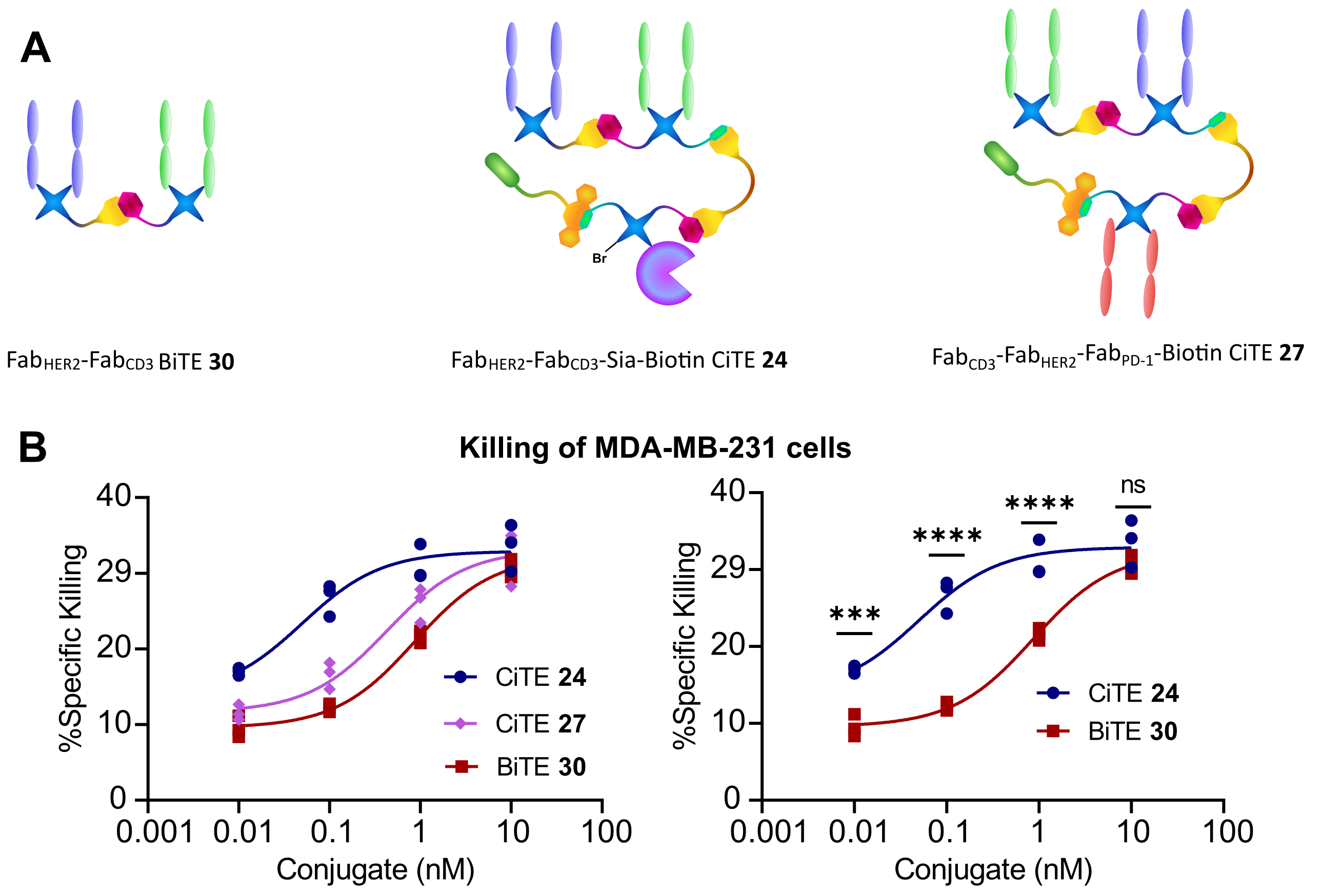
Figure 2 | Cytotoxicity assay of FabHER2‑FabCD3‑Sia‑Biotin CiTE 24 and FabCD3‑FabHER2‑FabPD‑1‑Biotin CiTE 27.
A | Structures of constructs used in assay. B | Cytotoxicity assay of FabHER2‑FabCD3‑Sia‑Biotin CiTE 24 and FabCD3‑FabHER2‑FabPD‑1-Biotin CiTE 27. MDA-MB-231 cells were co-cultured with T cells from a single donor (E:T ratio of 2:1) and treated with 0.01 – 10 nM of CiTE 24, CiTE 27 or BiTE 30. MDA-MB-231 viability was assessed 24 h following treatment via LDH assay. | Statistical analysis carried out with two-way ANOVA followed by post-hoc Tukey’s multiple comparisons test with multiplicity-adjusted P values with α = 0.05. *P < 0.05, **P < 0.01, ***P < 0.001, ****P <0.0001. Curves fitted with non-linear regression with the following model: [Agonist] vs response (three parameters).
References
- Robert, C. A decade of immune-checkpoint inhibitors in cancer therapy. Nat. Commun. 11, 10–12 (2020).
- Gera, N. The evolution of bispecific antibodies. Expert Opin. Biol. Ther. 22, 945–949 (2022).
- Szijj, P. & Chudasama, V. The renaissance of chemically generated bispecific antibodies. Nat. Rev. Chem. 5, 78–92 (2021).
- Maruani, A. et al. A Plug-and-Play Approach for the de Novo Generation of Dually Functionalized Bispecifics. Bioconjugate Chem. 31, 520–529 (2020).
- Thoreau, F. et al. Modular Chemical Construction of IgG-like Mono- and Bispecific Synthetic Antibodies (SynAbs). ACS Cent. Sci. 9, 476–487 (2023).
- Herrmann, M. et al. Bifunctional PD-1 3 aCD3 3 aCD33 fusion protein reverses adaptive immune escape in acute myeloid leukemia. Blood 132, 2484–2494 (2018).
- Gray, M. A. et al. Targeted glycan degradation potentiates the anticancer immune response in vivo. Nat. Chem. Biol. 16, 1376–1384 (2020).
Follow the Topic
-
Nature Chemistry
A monthly journal dedicated to publishing high-quality papers that describe the most significant and cutting-edge research in all areas of chemistry, reflecting the traditional core subjects of analytical, inorganic, organic and physical chemistry.





Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in