Benchmarking Single Cell RNA-seq: Two Complementary Approaches
Published in Bioengineering & Biotechnology
We actually would like to tell the story behind two papers, Ding et al. and Mereu et al. – each started independently and taking different paths towards the same goals. At the Broad Institute, early in 2017 a group came together to develop a plan to benchmark single cell RNA sequencing (scRNA-seq) experimental methods. The field had reached a point that there were many published protocols, but no clear consensus on the relative strengths and weaknesses of the methods. We realized that this would be important as the Human Cell Atlas (HCA) project was in its early stages of collecting data for this large-scale project. Meanwhile, Holger Heyn’s group at CNAG in Barcelona, involved in earlier benchmarking efforts, teamed up with many technology leaders of the HCA to develop a multi-center strategy for the benchmarking of the most commonly used scRNA-seq protocols with the view to define guidelines for the HCA and broader community.
In August, 2017, our two groups connected after Holger posted a message to the HCA Slack channel (#benchmark). We continued to work, coordinated on synergistic efforts, communicating regularly, and even had the Broad group contribute single nucleus RNA-seq to the CNAG study.
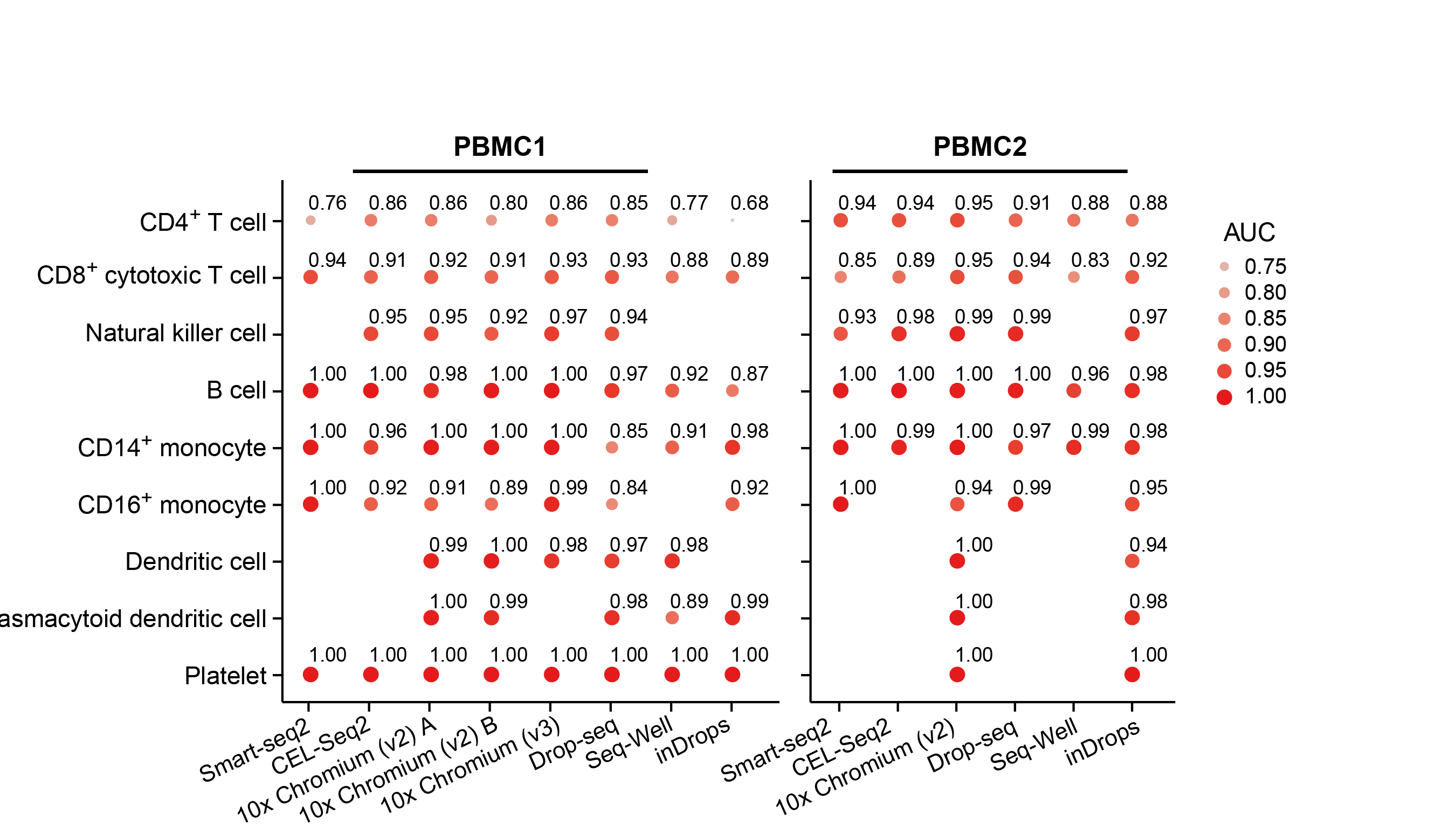
Our two groups took complementary approaches to benchmark these methods. The Broad group decided to utilize local expertise to process sample aliquots simultaneously and in parallel with each method. On the other hand, the CNAG group decided to create a large number of identical aliquots of a complex reference mixture of cells and send them to scRNA-seq experts all over the world to generate datasets. While the former was likely better controlled for some experimental variation, the latter was done by labs with extensive experience and expertise in each particular method. As is often the case, the computational analysis was complex and we faced many challenges – particularly trying to be fair to all methods. Each paper has its own analysis solutions – highlighting the scumi pipeline and AUC for cell type gene expression patterns from the Broad group and the matchSCore2 data projection workflow from the CNAG group.
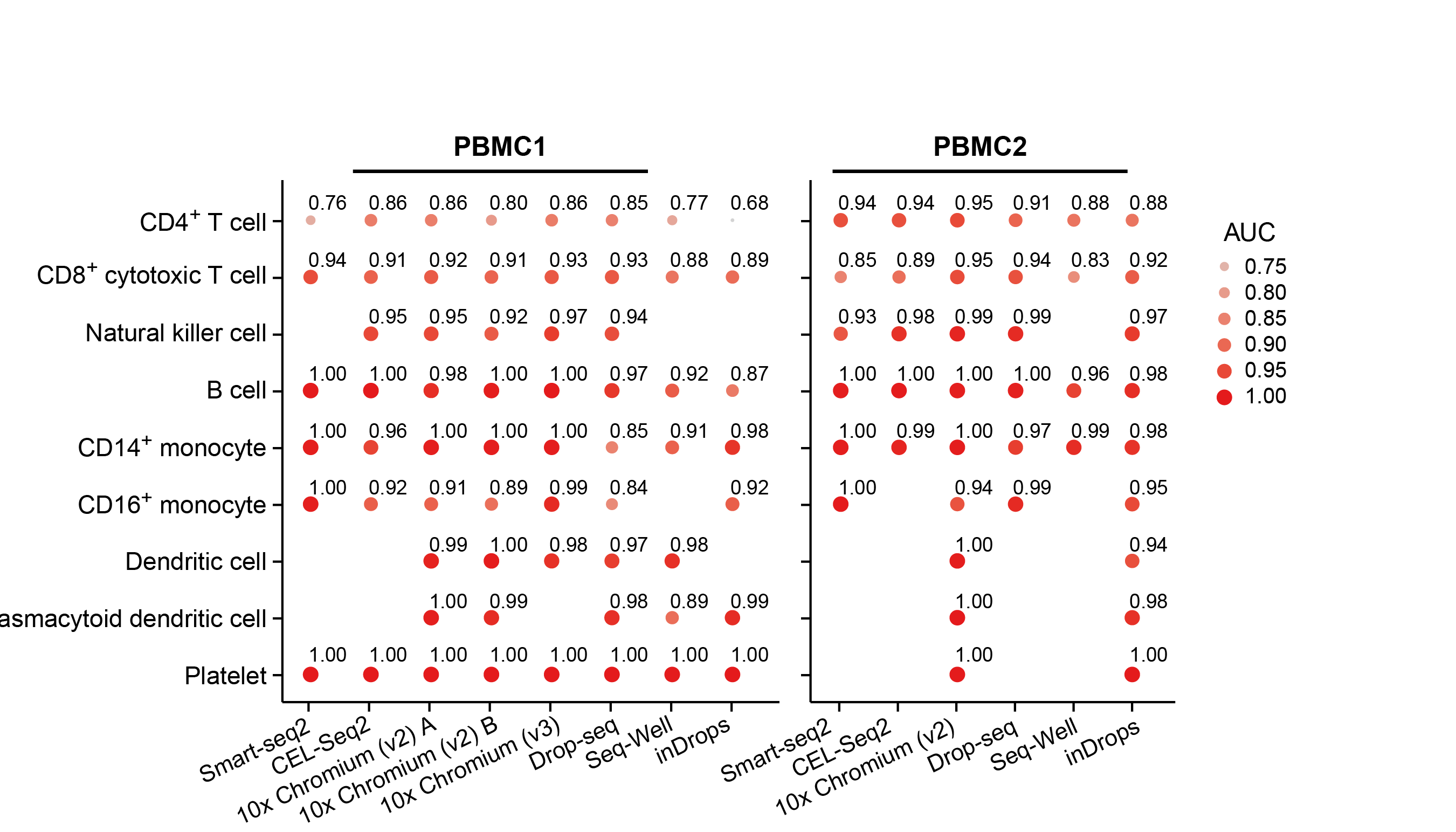
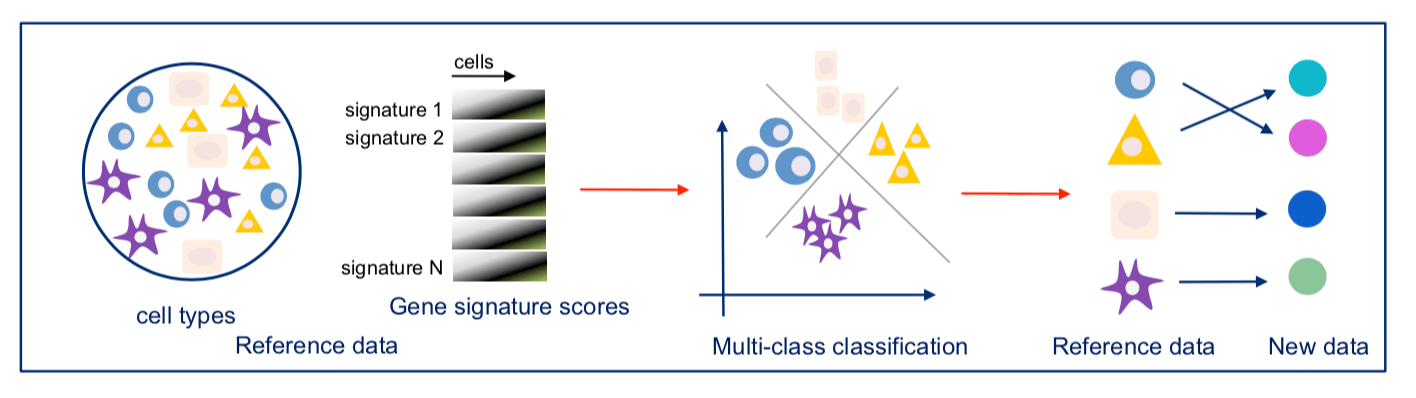
How do we envision these studies being valuable to the scientific community? Most immediately, these studies provide guidance to researchers on choosing among the available scRNA-seq methods. Additionally, they provide analysis tools and approaches to work jointly with different scRNA-seq datasets. Our sample selection should allow future studies, particularly those introducing new or improved methods, to make direct comparisons to our benchmark studies. Finally, what we did not fully understand when we started these projects is that these datasets are a highly valuable resource for those developing new computational approaches such as normalization, correction of batch effects, modeling and removing ambient RNA, and integration of experiments. We also hope that technical experts in other kinds of methods might consider adapting our benchmarking approaches to evaluate the array of methods in their own fields.
This blog post was a joint effort of the authors of both Nature Biotechnology papers.
Follow the Topic
-
Nature Biotechnology
A monthly journal covering the science and business of biotechnology, with new concepts in technology/methodology of relevance to the biological, biomedical, agricultural and environmental sciences.



Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in