Boosting dopamine tone rescues Parvalbumin interneuron deficits and hippocampal hyperexcitability in an Alzheimer's mouse
Published in Neuroscience and General & Internal Medicine
What we already knew:
The reduced excitability of Parvalbumin interneurons (PV-INs) is commonly associated with aberrant gamma-oscillations and with increased brain’s excitation/inhibition ratio. These deficits lead to subclinical epilepsy and brain hyperexcitability detectable in different neurological disorders, including Alzheimer’s Disease (AD). Indeed, experimental observations from AD animal models and AD patients proved a close association between reduced gamma-oscillations and cognitive impairments, highlighting that hyperexcitability can accelerate hallmarks of AD pathology and the onset of dementia. In fact, nowadays there is huge effort in rescuing the PV-IN activity or restoring gamma-waves in patients, paving the way for promising non-invasive treatments.
What was missing:
The question we wanted to answer was whether in AD the dysfunctions in hippocampal PV-INs could be due to (or at least associated with) a drop in the dopaminergic tone in this area. In fact, our journey began years ago with the captivating discovery that the dopamine neurons in the Ventral Tegmental Area (VTA) degenerate precociously in AD, leading to a drop of dopamine in VTA-projecting areas - such as the hippocampus - and to the onset of several cognitive and non-cognitive deficits. Subsequent clinical research confirmed that dysfunctions in the mesocorticolimbic dopamine system are prodromal factors that speed up the progression of AD and contribute to cognitive and neuropsychiatric symptoms.
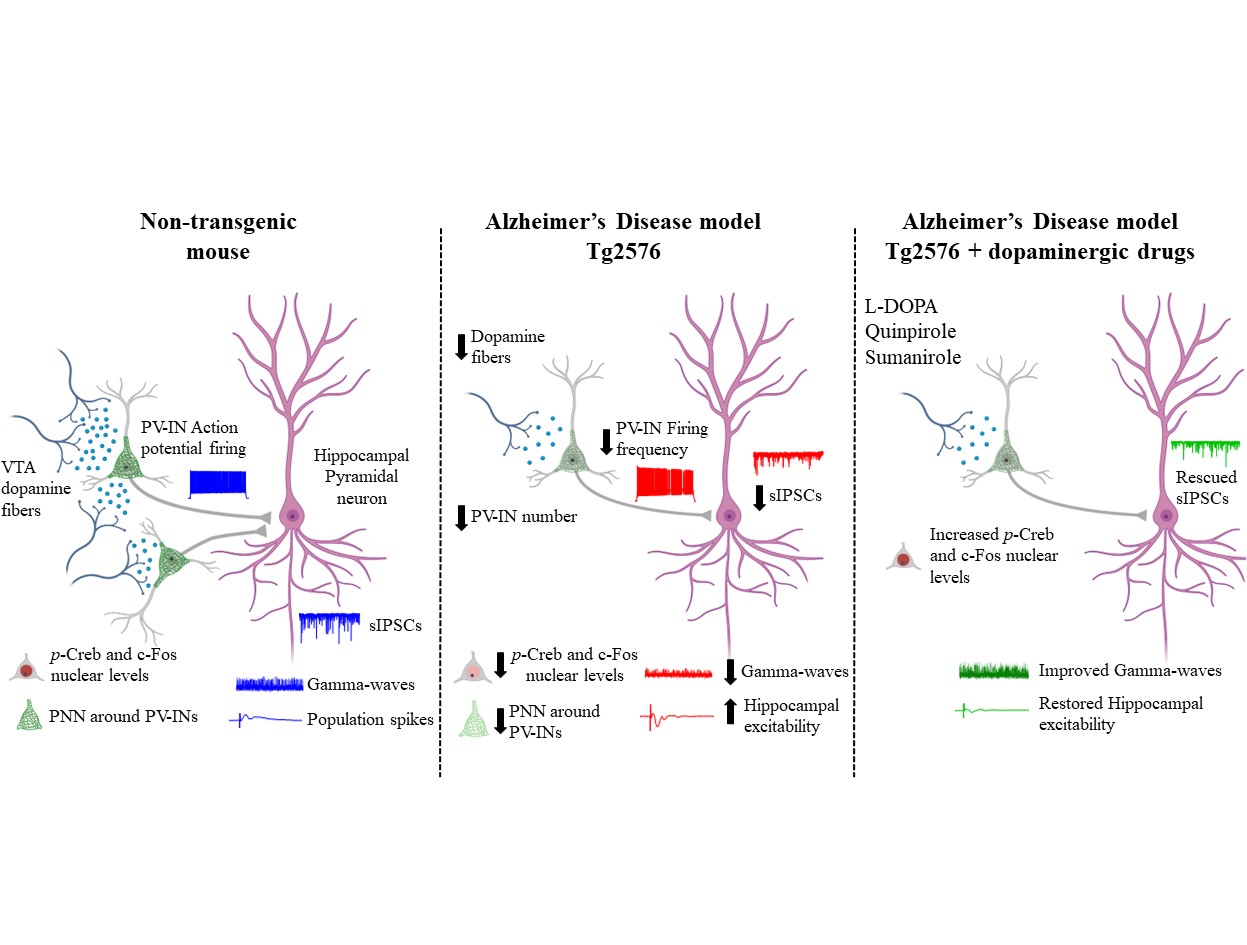
In this context, we engaged in intriguing research to prove that the precocious dopamine neuron loss in the VTA leads to hippocampal PV-IN dysfunctions and to brain hyperexcitability in the Tg2576 mouse model of AD.
What we did and what we discovered:
We adopted a multidisciplinary approach, ranging from slice electrophysiology to stereology and confocal microscopy, to prove that the reduced VTA dopamine signaling in the Tg2576 hippocampus affects the functioning of PV-INs, causing network hyperexcitability.
We began by confirming the reduced dopaminergic input directly on PV-INs, by injecting the AAV1-hSyn-FLEx-mGFP-2ASyPhy-mRuby virus in the VTA of DAT-Cre crossed with Tg2576 mice, to label selectively the dopamine neurons (with mGFP) and their presynaptic inputs containing synaptophysin (with mRuby).
However, whether this reduced dopaminergic input onto PV-INs could be responsible for defects of PV-IN function and survival remained an open question. Thus, we went ahead to provide some intriguing evidence pointing to this direction.
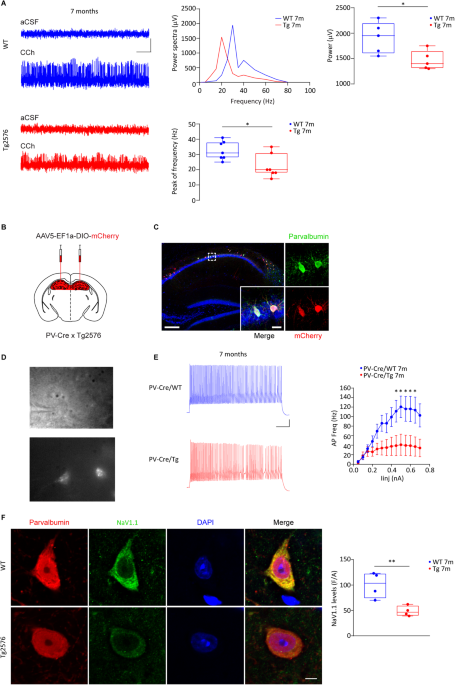
We first wondered if the excitability of PV-INs in the hippocampus of Tg2576 mice was reduced. Therefore, we recorded from PV-INs in the CA1 region of Tg2576 mice and confirmed an important rundown in their ability to fire action potentials, associated also with lower expression of NaV1.1 – the principal sodium channel in GABAergic neurons, responsible for action potential generation and propagation. Given the reduced excitability of Tg2576 PV-INs, we then asked whether the CA1 pyramidal neurons received a weaker inhibitory tone that could lead to a shift towards hyperexcitability in the Tg2576 hippocampus. Indeed, we observed reduced spontaneous inhibitory currents and increased number of population spikes. Finally, we also showed that Tg2576 mice were affected by a reduction in the power and peak frequency of gamma-oscillations, proving an impairment in the hippocampal oscillatory circuit. Most importantly, our analysis revealed that the GABAergic drive onto CA1 pyramidal neurons was unaffected at an age before the VTA dopamine neurodegeneration, suggesting a direct connection between dopamine loss and GABAergic deficits.
By further investigating the PV-IN deficits in Tg2576 mice, we stumbled upon another intriguing observation: the number of hippocampal PV-INs was reduced in Tg2576 mice. Additionally, the perineuronal nets (PNNs) that surround these neurons and ensure synaptic stabilization and neuronal protection were reduced compared to age-matched control mice, likely making PV-INs of Tg2576 mice more vulnerable. In line with a link between PV-IN survival and dopamine neuron loss, no changes in PV-IN number were evident before the VTA degeneration, strengthening the notion that dopamine could be necessary for PV-IN survival.
To delve deeper into our exploration, we analyzed the CREB/c-Fos pathway in PV-INs as it is involved both in neuronal survival and activity and is downstream of dopamine receptors. Coincident with the VTA dopamine neurodegeneration, we observed reduced nuclear levels of both p-CREB and c-Fos in PV-INs of Tg2576 mice. This result is in line with the notion that the reduced dopaminergic drive in PV-INs could be responsible for the decreased activation of the CREB/c-Fos pathway, thus affecting the survival and function of these neurons.
Does a dopamine boost work?
To directly prove that the loss of dopamine from the VTA drives the observed PV-IN dysfunctions, we next performed different pharmacological treatments to modulate the dopaminergic signaling.
Specifically, we found that the selective D2-like receptor agonist quinpirole increased p-CREB and c-Fos levels in Tg2576 PV-INs. Accordingly, the D2-like receptor antagonist sulpiride reduced p-CREB levels in WT PV-INs, mimicking Tg2576 features. Additionally, L-DOPA enhanced the inhibitory drive on CA1 pyramidal neurons and ameliorated hippocampal hyperexcitability.
What makes these findings more exciting is the potential of dopaminergic drugs to counteract AD progression, carrying significant clinical implications. In line with this, the highly selective D2-receptor agonist sumanirole – an effective anticonvulsant in animal models of seizures – increased p-CREB levels and enhanced gamma-oscillations in Tg2576 mice.
Overall, our data prove that boosting the dopaminergic signaling is sufficient to rescue hippocampal PV-IN dysfunction, reduced gamma-oscillations and brain hyperexcitability.
Our conclusion
Our study hints at the VTA dopaminergic neuron loss as a crucial event triggering the insurgence of epileptic-like activity since the early phases of AD. We added valuable insights into the mechanisms at the basis of hippocampal network defects. Most importantly, our work paves the way for the precocious administration of dopaminergic drugs to protect PV-INs from dysfunctions, thus preventing brain hyperexcitability and slowing down of AD progression, hopefully offering a new prospect for future therapeutic approaches.
Follow the Topic
-
Molecular Psychiatry
This journal publishes work aimed at elucidating biological mechanisms underlying psychiatric disorders and their treatment, with emphasis on studies at the interface of pre-clinical and clinical research.



Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in