
How do we predict chemical reaction selectivity, and how can we get those answers from molecular modeling? These were the questions I was faced with during my PhD in the late 1980’s. To make it even more difficult, we wanted to predict asymmetric transition metal catalyzed reactions that were notoriously hard to model. In the mid-1990’s, we had some experience with what could and could not be done. We knew that isomeric ratios can be obtained from calculated energies. Modeling an asymmetric selectivity requires you to account for effects in the transition state (TS); only there will the isomeric paths show an energy difference that corresponds to the reaction selectivity.
At that time, problems like asymmetric catalysis simply could not be directly addressed by quantum mechanics (QM). Density Functional Theory (DFT) was making rapid advances, but we knew that the critical factor to differentiate enantiomeric products would be non-bonded interactions between substituents of the substrate and catalyst in the transition state. Important interactions such as London dispersion simply were not available from DFT, or any other QM methods that were practical for our target systems. However, we had experience with molecular mechanics (MM), where some of the available methods had been tuned specifically to give reliable nonbonded interactions. We had already shown that MM methods could be parameterized to give accurate structures and energies of intermediates in transition metal catalyzed reactions.
We were not the first ones to think of MM in this context. Transition state force fields (TSFF) were popularized in the 1980’s, but the practice of training such models from experimental data led to very underdetermined parameter sets with low transferability. It had been shown in the 1980’s that standard force fields could be derived from quantum mechanical (QM) data, by using QM determined vibrations to train the necessary force constants in MM, but it was not clear how this could be applied to TSFFs. Transition states are saddle points, where one of the many reaction coordinates has a negative curvature, giving a negative force constant. For our intended use, the TSFF should have a positive curvature in all directions, to allow us to find transition states by simple downhill search algorithms.

Picture credit: Taylor Quinn (@tayquanderoga) and Tony Rosales (@trrosales)
The transformative moment is one I still remember well some 22 years later. I was out walking an early morning when inspiration struck. The QM vibrational data will define the shape of the TS in all directions by a set of eigenvectors and eigenvalues, one of which is negative. By simply changing the value from negative to strongly positive, we could create a new surface, with structure and vibrations exactly the same as for the TS, except for along the reaction coordinate where we’ve now introduced an energy increase in both directions. This allowed us to train the MM method using the modified QM data, in effect restricting all low energy structure to the TS region. The beauty of the approach is that all small distortions of the TS that are not along the reaction coordinate will be correctly reproduced, giving good energies for distorted transition structures even if they are no longer saddle points. From this basic idea, we could create force field models of arbitrary TSs. We called this method “quantum to molecular mechanics”, shortened “Q2MM”.
We showed that we could use very small TS models, easily affordable by DFT even two decades ago, and use these to successfully train TSFFs. For anything more than 3 bonds away from the breaking and forming TS bonds, we used traditional ground state force fields, selected for the internal balance. By doing extensive conformational searches, we could find all possible forms of the TS (that is, all reaction paths), and the energies of these agreed very well with experimentally observed selectivities.
Fast forward two decades, we have shown that without training from experiments, we can calculate experimental selectivities, with an accuracy over large sets of diverse structures rivalling any other method in use today. In short, this is possible because so far, QM methods that are accurate enough to get all the non-bonded interactions right are computationally too expensive to allow searching through all possible reaction paths. Other force field methods are fast enough, but in general have not been trained to reproduce energies at transition states.
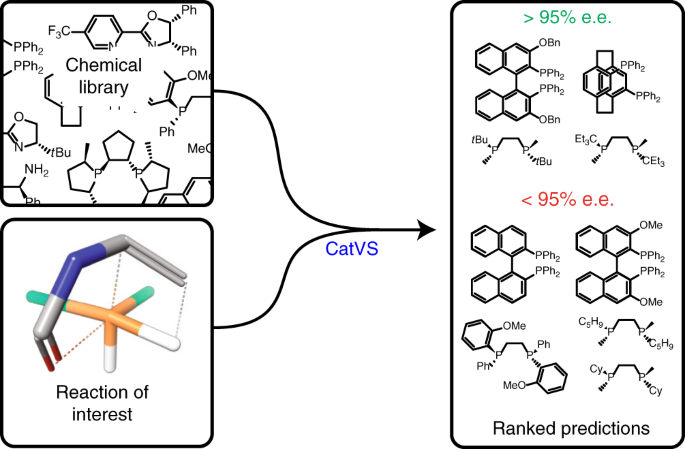
We have now applied the method to virtual screening of asymmetric catalysts in an industrial setting. We developed web interfaces that allow a chemist to draw their desired substrate, and then submit it for screening. The program will automatically identify the reactive moiety, combine it with a library of catalysts, search for all possible conformations, and send back a list of expected stereoselectivities. The method was applied to two reactions developed by recipients of the 2001 Nobel Prize for asymmetric catalysis. We got very significant enrichment in catalyst selection. If you are interested in the results, or want to find references to what has gone before, or in limitations of the method (which do exist), you can read our article in Nature Catalysis:
“Rapid Virtual Screening of Enantioselective Catalysts Using CatVS”, Anthony R. Rosales, Jessica Wahlers, Elaine Limé, Rebecca E. Meadows, Kevin W. Leslie, Rhona Savin, Fiona Bell, Eric C. Hansen, Paul Helquist, Rachel H. Munday, Olaf Wiest, and Per-Ola Norrby, Nature Catal. http://dx.doi.org/10.1038/s41929-018-0193-3.
I’m very grateful to my coauthors and other collaborators who have supported this project over decades, in particular: Paul Helquist and my PhD supervisor Björn Åkermark who threw this problem at me in 1986; and Olaf Wiest who now continues the academic Q2MM development.
Follow the Topic
-
Nature Catalysis
This journal brings together researchers from across all chemistry and related fields, publishing work on homogeneous catalysis, heterogeneous catalysis, and biocatalysts, incorporating both fundamental and applied studies.





Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in