Catching recombination in the act: the rewards of large-scale COVID-19 monitoring
Published in Microbiology, Protocols & Methods, and Biomedical Research
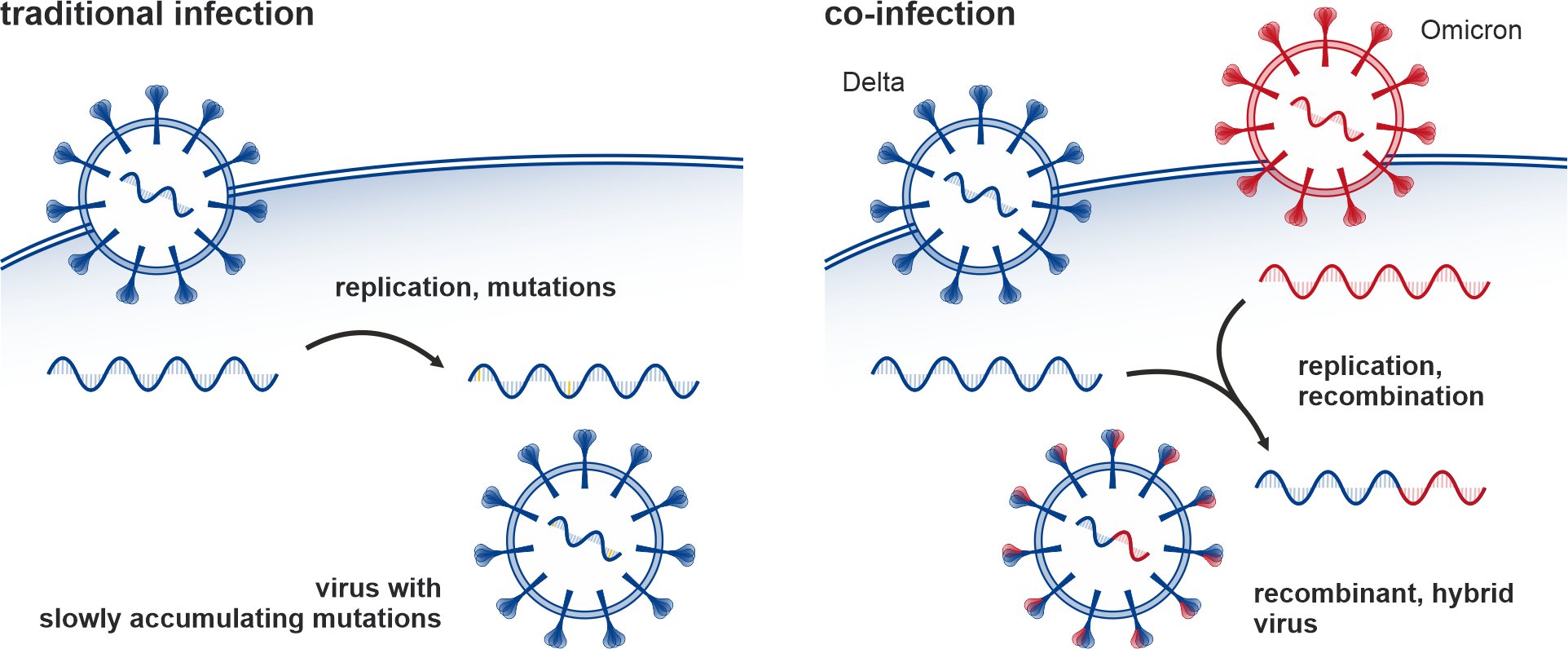
It all starts with the Data
Besides the terrors of a global pandemic, the emergence of COVID-19, caused by the SARS-CoV-2 coronavirus, has brought about a hitherto unprecedented effort in international cooperation and worldwide monitoring. Since 2019, a massive amount of genomic viral data has accumulated and been made publicly available on the Internet.
The Versatile Emerging infectious disease Observatory project (VEO, https://www.veo-europe.eu), with a kick-off date of January 2020, originally aimed to provide actionable information on a wide variety of emerging diseases and to promote early-warning using a versatile, interdisciplinary approach. However, with the rapid spread of COVID-19 starting from the end of 2019, much of the collaboration’s initial efforts have naturally been directed towards SARS-CoV-2 monitoring and surveillance.
Among various achievements in the field, the consortium developed a standardised workflow to uniformly analyse raw SARS-CoV-2 next-generation sequencing data deposited in the European Nucleotide Archive (EBI-ENA, https://www.ebi.ac.uk/ena/browser/home) by an enormous number of researchers and institutions worldwide. A searchable database (European COVID-19 Data Portal, https://www.covid19dataportal.org/) was created containing the mutations with their genomic positions, allele frequencies and additional metadata of the samples. The database is unique in the sense, that it contains the minor (low alternate allele frequency) variations in the samples in addition to the consensus sequences (which are available in the widely used GISAID Database, https://gisaid.org/).
Big data – minor variants
Given that emerging variants/lineages might circulate as minor ones initially before spreading, comprising only a small fraction of the viral population in sequenced samples, the European COVID-19 Data Portal and the subsequently created CoVEO database containing low alternate allele frequency mutations can be utilized to detect these instances. Moreover, these datasets make it possible to search for the unique markers of different variants (e.g. Alpha, Delta, Omicron) simultaneously within a sample, to identify dual infection or co-infection cases (see Figure 1a-b). Co-infections may increase the severity of the disease (although not necessarily), furthermore, the genome of the two variants may combine during replication, resulting in a new, hybrid variant, with a recombinant genome. In the study, our objective was to detect co-infection cases in this vast dataset of over 2 million SARS-CoV-2 samples collected globally and find signs of recombination within them. While co-infection cases were reported earlier from various countries (studying a few thousand samples each), a comprehensive worldwide analysis of this scale had not been conducted before.
 infection, the genome of the virus might acquire mutations during viral replication, creating a largely similar, but slightly modified virus. b. During co-infection, the genomes of the two infecting viruses might mix in the course of replication, producing an entirely new, hybrid virus. c. The timeline of Delta and Omicron occurrences (top, coloured curves) alongside the detected Delta – Omicron co-infection cases (bottom, vertical lines).")
Dual infections: how rare are they and why do they matter?
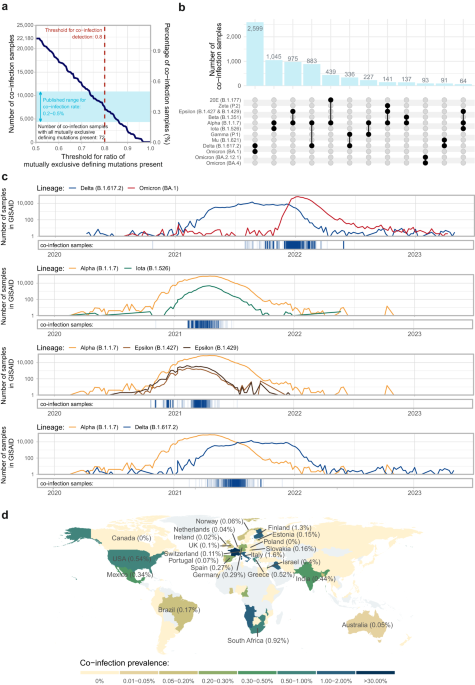
Due to the relatively high mutation rate of the SARS-CoV-2 coronavirus, several variants emerged and spread since its first appearance in Wuhan. These diverse variants have unique defining mutation combinations, however, the number of these markers varies depending on the variant: some have only two, while others have up to twenty. To increase the statistical power of our analysis, we expanded the list of these variant indicators by including mutually exclusive defining mutations in specific variant combinations.
We have found convincing signs of co-infection (i.e. the simultaneous presence of the vast majority of defining mutations of more than one variant) in 0.35% of the samples (7700 cases), in most cases Delta-Omicron combinations. Naturally, co-infections generally occurred in large numbers when the comprising variants were concurrently present and spreading in the investigated population. For an illustration, see the timeline of Delta – Omicron co-infection cases in Figure 1c. Co-infection samples (shown with blue vertical lines in the bottom panel) were common from the end of 2021 until the start of 2022, exactly in the time period when both traditional Delta and Omicron infections appeared in large numbers worldwide (blue and red curves in the above panel).
Out of all countries, South Africa exhibited the highest co-infection rate, where the immunosuppressed population is relatively high due to the high number of untreated patients with AIDS caused by the HIV virus. Immunosuppression might prolong infections, leading to more accumulated mutations caused by genetic drift, which effect might have contributed to the appearance of the Omicron variant at the end of 2021. Additionally, prolonged infection increases the likelihood of an individual being concurrently infected with another variant, and the weakened immune system leads to accelerated virus replication, potentially resulting in recombination, given that it happens usually during the replication of the virus genome via template-switching, when RNA-dependent RNA-polimerase accidentally drops from one variant’s genome to another’s.
Real-time or virtual recombination?
Viral variants of a recombinant origin have been recently circulating in large numbers, in fact, all viral sequences assigned to a Pango lineage whose prefix starts with an “X” (e.g. the currently spreading XBB variants) belong to recombinant lineages (or their non-recombinant descendants), comprising about 2% of the total publicly available GISAID dataset. However, prior to the transmission and subsequent spread of a recombinant genome, a recombination event must take place within a single host co-infected by the non-recombinant parental lineages. If recombination occurs in a co-infection sample, traces of the recombinant genome might be detectable in a small fraction of the sequenced viral population. However, the identification of such a scant genomic subpopulation is hampered by multiple factors. For instance, the distribution of defining mutations is not uniform along the genome, most of them are located on the S gene (coding the Spike protein), which makes it virtually impossible to detect recombination breakpoints in genomic regions, where the defining mutations of the two variants are scarce. Moreover, chimeric reads formed during the PCR amplification phase of amplicon sequencing are indistinguishable from real recombinant reads.
Keeping these sources of ambiguity in mind, we aimed to develop a pipeline, which can detect intra-host recombination real-time, before the resulting recombinant genome spreads, for which we implemented two different approaches.
Where do we go from here?
Our results and methodology highlight the benefits and scientific potential of organized worldwide surveillance of pathogens. The never-before-seen sequencing output produced during the COVID-19 pandemic has allowed us to systematically examine millions of samples in search of dual infections and real-world in-situ recombination events. The algorithmic pipeline outlined in our paper can help monitor and forecast important milestones in viral evolution and might also serve as a stepping stone for other large-scale genomic studies of viruses. With the advent of the increasingly popular long-read sequencing technologies, our analysis approach may further be refined for more precise recombinant detection.
Our research has received funding from the European Union's Horizon 2020 research and innovation programme under grant agreements No. 874735 (VEO) and No.101046203 (BY-COVID).
Follow the Topic
-
Nature Communications
An open access, multidisciplinary journal dedicated to publishing high-quality research in all areas of the biological, health, physical, chemical and Earth sciences.

Related Collections
With Collections, you can get published faster and increase your visibility.
Women's Health
Publishing Model: Hybrid
Deadline: Ongoing
Tumor Microenvironment Crosstalk and Therapeutic Implications
Publishing Model: Hybrid
Deadline: Nov 02, 2026


Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in