Caught in a Folate Trap: Killing Cancer Cells by Inhibiting MTHFD1
Published in Cancer
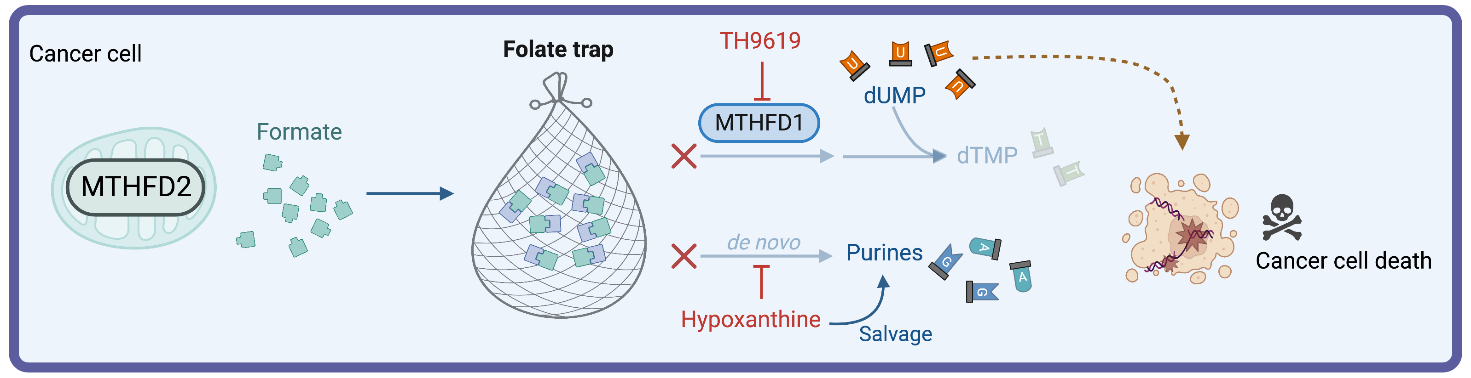

Where did we start from
The targeting of one-carbon metabolism (1C) in cancer cells is historically well established with anti-folates applied in clinical practice. However, current anti-folate therapy negatively affects healthy cells, causing a range of side effects. Studies showed that 1C enzymes and especially mitochondrial methylenetetrahydrofolate dehydrogenase/cyclohydrolase (MTHFD2) are highly upregulated in cancer cells compared to normal tissue.1 We found that in addition to proliferation, the mitochondrial part of 1C is crucially involved in cancer cell motility and metastasis.2
Thus, we developed and described inhibitors that target specifically MTHFD1/2, namely TH9619 and TH9975 (MTHFDi).3 In the current study, we joined forces in a collaborative approach between the University of Sheffield, Karolinska Institute and the Luxembourg Institute of Health to deepen our mechanistic understanding of the inhibitors’ mode of action.
TH9619 does not inhibit mitochondrial MTHFD2, but is most toxic in MTHFD2 overexpressing cancer cells
When we began to study the metabolic consequences of MTHFDi, we realized that our prior understanding of the inhibitors’ mode of action was incomplete. From our earlier studies, we learned that both compounds efficiently bind to MTHFD1 and MTHFD2 in biochemical assays and that deleting MTHFD2 conferred resistance.
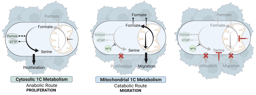
Yet, when we measured formate release from cells, we found that, in contrast to MTHFD2-/- cells, MTHFDi-treated cells still released formate in comparable rates to untreated cells (Figure 1A). As formate release is a direct readout of mitochondrial 1C activity (Figure 1B),4,5 this indicates that mitochondrial 1C is still active in MTHFDi-treated cells.
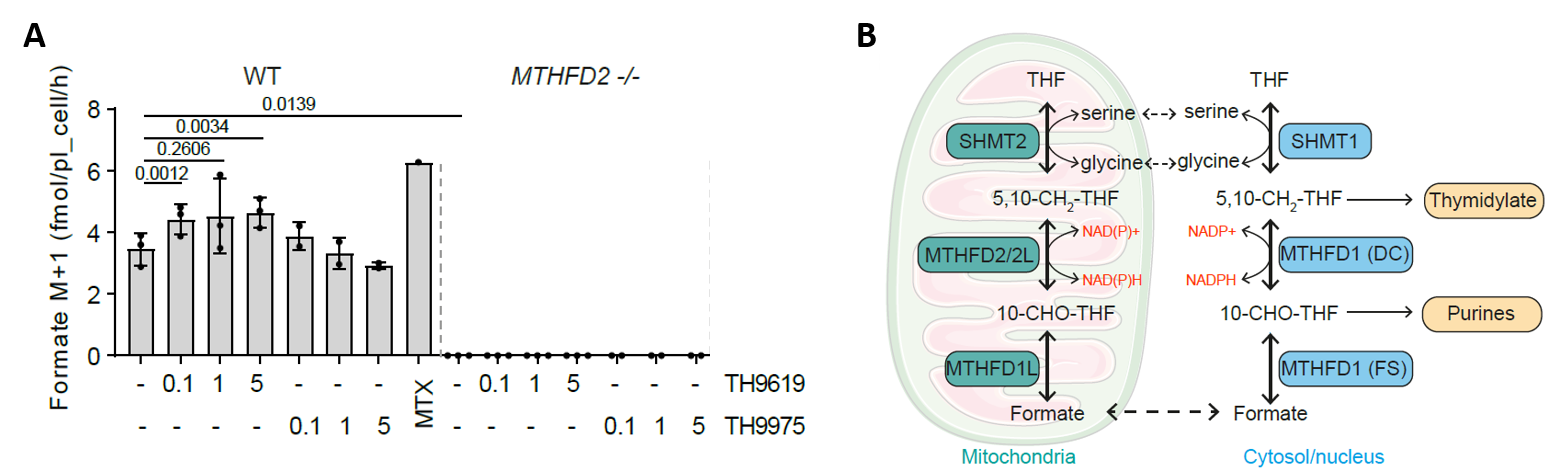
This made us wonder why the inhibitors were most toxic in cells with high MTHFD2 expression, while our data clearly showed that mitochondrial MTHFD2 itself was not inhibited in cells.
TH9619 has unique anti-cancer effects in MTHFD2+/+ and MTHFD2-/- cells
Our next step was to understand what happens to nucleotide synthesis in MTHFDi-treated cells in dependence of MTHFD2 expression. Measurements of nucleotide levels and synthesis and metabolite rescue experiments revealed that the impact of MTHFDi on 1C is distinctly different between MTHFD2+/+ and MTHFD2-/- cells.
In MTHFD2+/+ cells, thymidylate and purine synthesis was severely depleted, but rescue experiments told us that the primary toxicity was due to depletion of thymidylate (Figure 2A). This contrasted with MTHFD2-/- cells, which were not rescued by thymidine and showed a complete block in purine synthesis (Figure 2B). These different nucleotide synthesis defects altered the biological response of the cells. In MTHFD2-/- cells, proliferation was halted but the cells did not die, whereas MTHFD2+/+ cells were being killed by MTHFDi.
Thus, purine depletion was cytostatic, but only thymidylate depletion, which causes uracil misincorporation into DNA, is actually cytotoxic to MTHFDi-treated cancer cells.
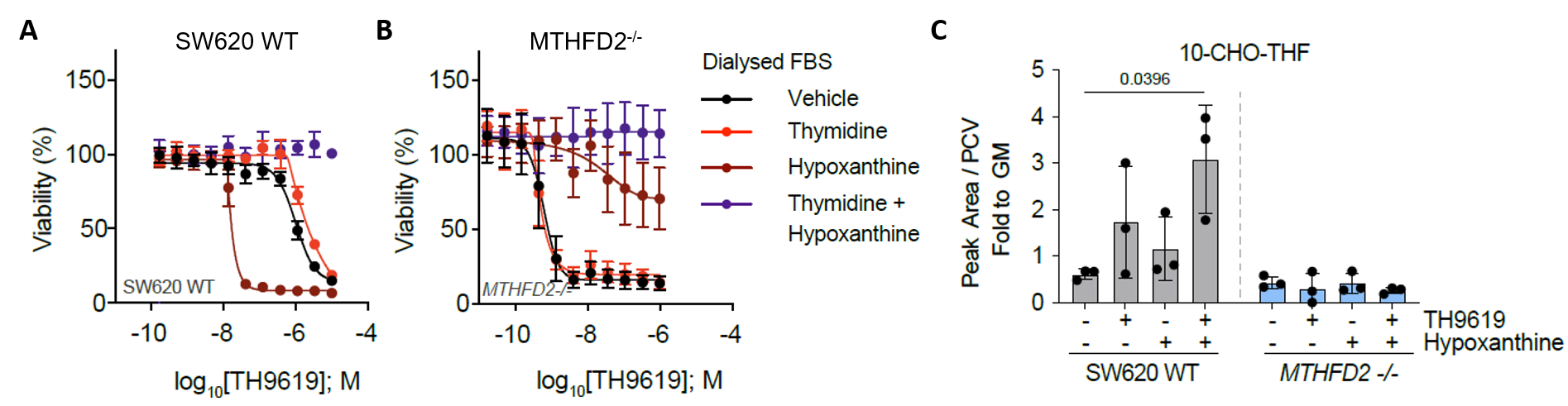
We discovered that hypoxanthine potentiated MTHFDi. Initially, we thought this was because hypoxanthine was being converted to the UMPS-inhibitor uric acid, but this turned out not to be the case.
Instead, hypoxanthine activated purine salvage, and thus acted as a feedback inhibitor of de novo synthesis of purines. In consequence, the intermediate 10-formyl-tetrahydrofolate (10-CHO-THF) is not consumed to make purines and cannot be used by MTHFD1 for synthesis of thymidylate (Figure 1B). We thus proposed that 10-CHO-THF was essentially trapped with no way to be consumed and that mitochondrial formate overflow would lead to 10-CHO-THF accumulation (Figure 3).
However, measuring folate intermediates is inherently challenging due to their low abundance, interconversion and versatile polyglutamylation. These analyses were challenging, they required us to substantially upscale experiments and to establish derivatization and LC-MS analysis protocols. After months of troubleshooting, we were excited to validate our hypothesis that MTHFDi cause folate trapping in cancer cells (Figure 2C).
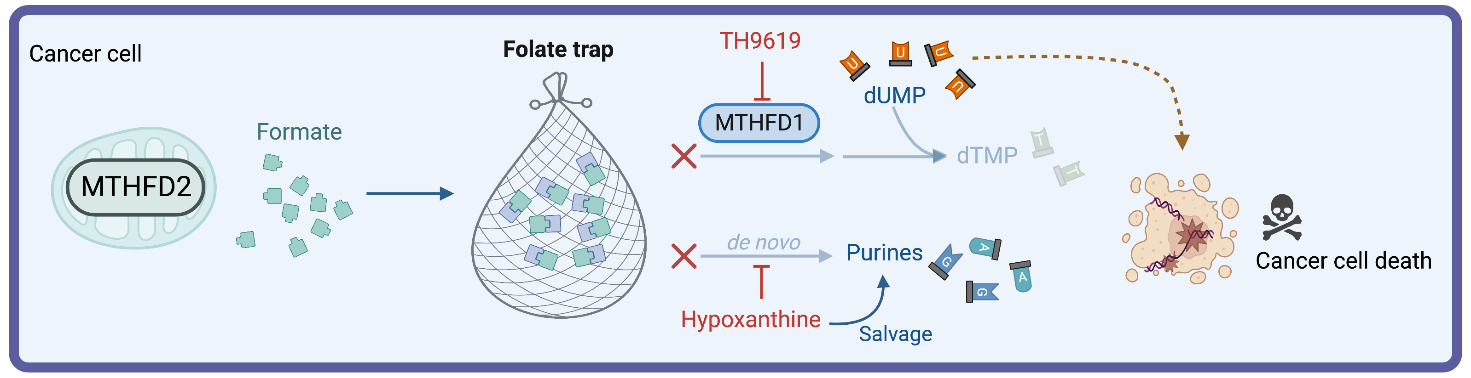
Perhaps not surprisingly, this folate trap was not generated in MTHFD2-/- cells, because they do not have formate overflow to feed the folate trap. However, we managed to induce a folate trap in MTHFD2-/- cells with formate and hypoxanthine addition which induced toxic thymidylate depletion, resulting in apoptosis rather than proliferation arrest.
Outlook and Beyond
Based on our increased understanding of the mode of action of MTHFDi, we are now interested in developing MTHFDi towards the clinic. Yet, some challenges remain and predicting MTHFDi efficacy in humans will be difficult. In mice, plasma thymidine and folate levels are much higher than in humans, whereas hypoxanthine levels are low. As these metabolites control drug efficacy, mice poorly model human physiology. Our future research will focus on using patient material to better recapitulate human physiology and on finding MTHFDi biomarkers for sensitivity.
About us
Luxembourg Institute of Health
The Luxembourg Institute of Health is a public biomedical research organization focused on precision health. The Cancer Metabolism Group at LIH’s Department of Cancer Research aims to characterize metabolic liabilities of cancer cells in their specific microenvironment to identify metabolic vulnerabilities for the development of new therapeutic concepts.
The University of Sheffield
The University of Sheffield is one of the world’s top 100 universities. Located in the Department of Oncology and Metabolism, the Cancer and Bone Laboratory researches molecular and metabolic mechanisms that govern tumorigenic processes, and exploits vulnerabilities for therapeutic development.
Science for Life Laboratory, Karolinska Institute
Karolinska Institute is one of the world’s leading medical universities. As part of the Department of Oncology and Pathology, the Helleday Laboratory is based at SciLifeLab. The research efforts in the Helleday laboratory are aimed at unraveling and harnessing defects in the DNA damage response and cell metabolism to develop novel therapies targeting diseases such as cancer.
References
1 Nilsson, R. et al. Metabolic enzyme expression highlights a key role for MTHFD2 and the mitochondrial folate pathway in cancer. Nat Commun 5, 3128, (2014).
2 Kiweler, N. et al. Mitochondria preserve an autarkic one-carbon cycle to confer growth-independent cancer cell migration and metastasis. Nat Commun 13, 2699, (2022).
3 Bonagas, N. et al. Pharmacological targeting of MTHFD2 suppresses acute myeloid leukemia by inducing thymidine depletion and replication stress. Nat Cancer 3, 156-172, (2022).
4 Meiser, J. et al. Increased formate overflow is a hallmark of oxidative cancer. Nat Commun 9, 1368, (2018).
5 Meiser, J. et al. Serine one-carbon catabolism with formate overflow. Sci Adv 2, e1601273, (2016).
Illustrations were prepared with BioRender.
Follow the Topic
-
Nature Metabolism
This journal publishes work from across all fields of metabolism research that significantly advances our understanding of metabolic and homeostatic processes in a cellular or broader physiological context, from fundamental cell biology to basic biomedical and translational research.

Related Collections
With Collections, you can get published faster and increase your visibility.
The expanding therapeutic landscape of GLP 1 receptor agonists
Publishing Model: Hybrid
Deadline: Jan 23, 2027
Microbiome and energy metabolism
Publishing Model: Hybrid
Deadline: Dec 06, 2026
Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in