Chemical biology approaches lead to discovery of a new protein lipidation regulatory enzyme
Published in Chemistry
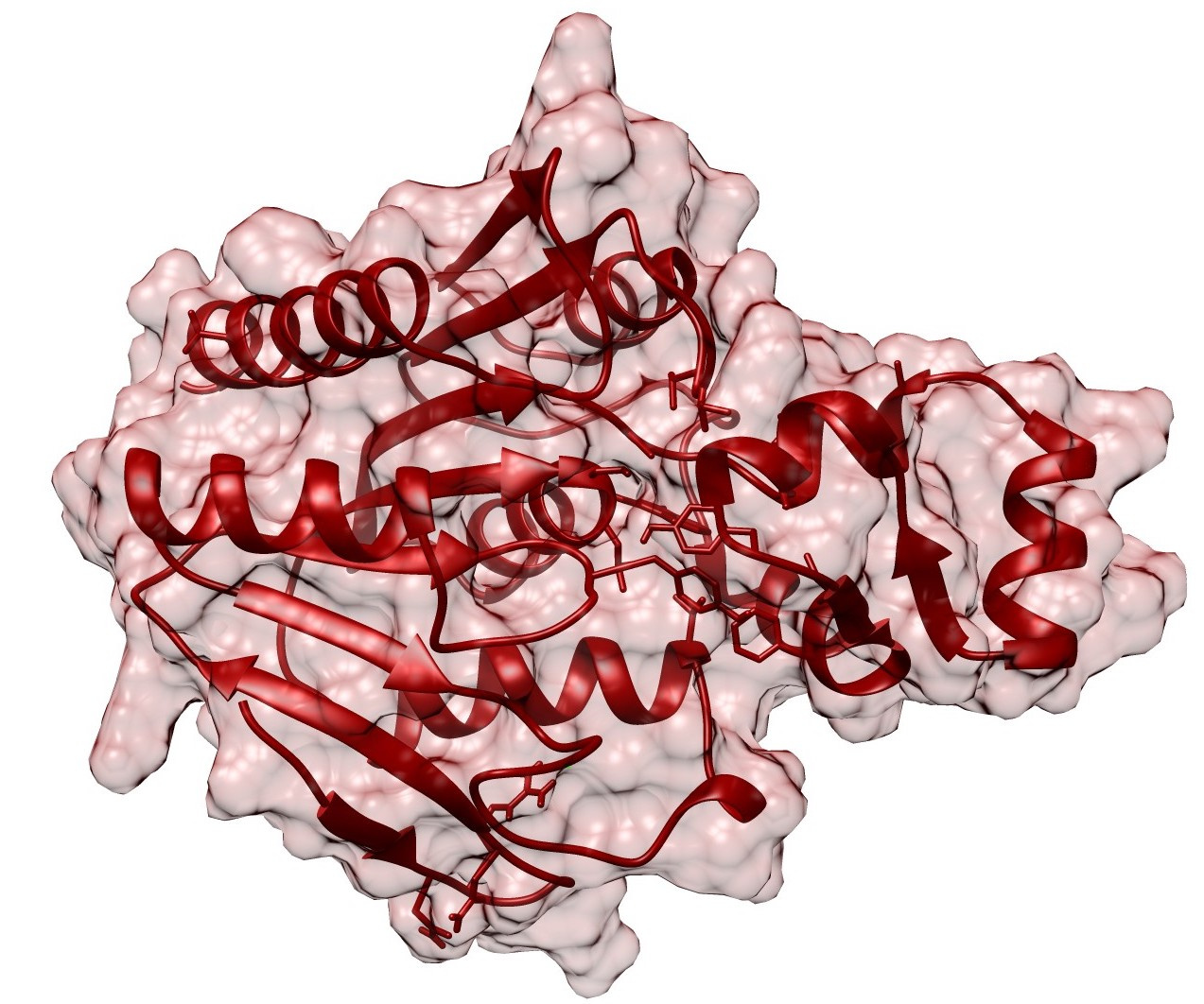

Dynamic protein lipidation through the modification of cysteine residues with thioesters of fatty acids provides a mechanism to tune proteome-wide properties such as hydrophobicity and membrane interactions. Analogous to kinase and phosphatase-based phosphorylation signaling, protein "S-acylation" (also referred to as "S-palmitoylation" since palmitate is the most abundant lipid post-translational modification (PTM) used in this pathway) is modulated by dedicated "writer" (acyl transferase) and "eraser" (deacylase) enzymes. Although thousands of human proteins are subject to modification by S-acylation PTMs, there are only 6 annotated "erasers" for this mark, named acyl-protein thioesterases (APTs). A key question my lab has been trying to investigate over the past several years is how this PTM is regulated, with the hypothesis being the spatial and temporal regulation are critical aspects of this signaling pathway.
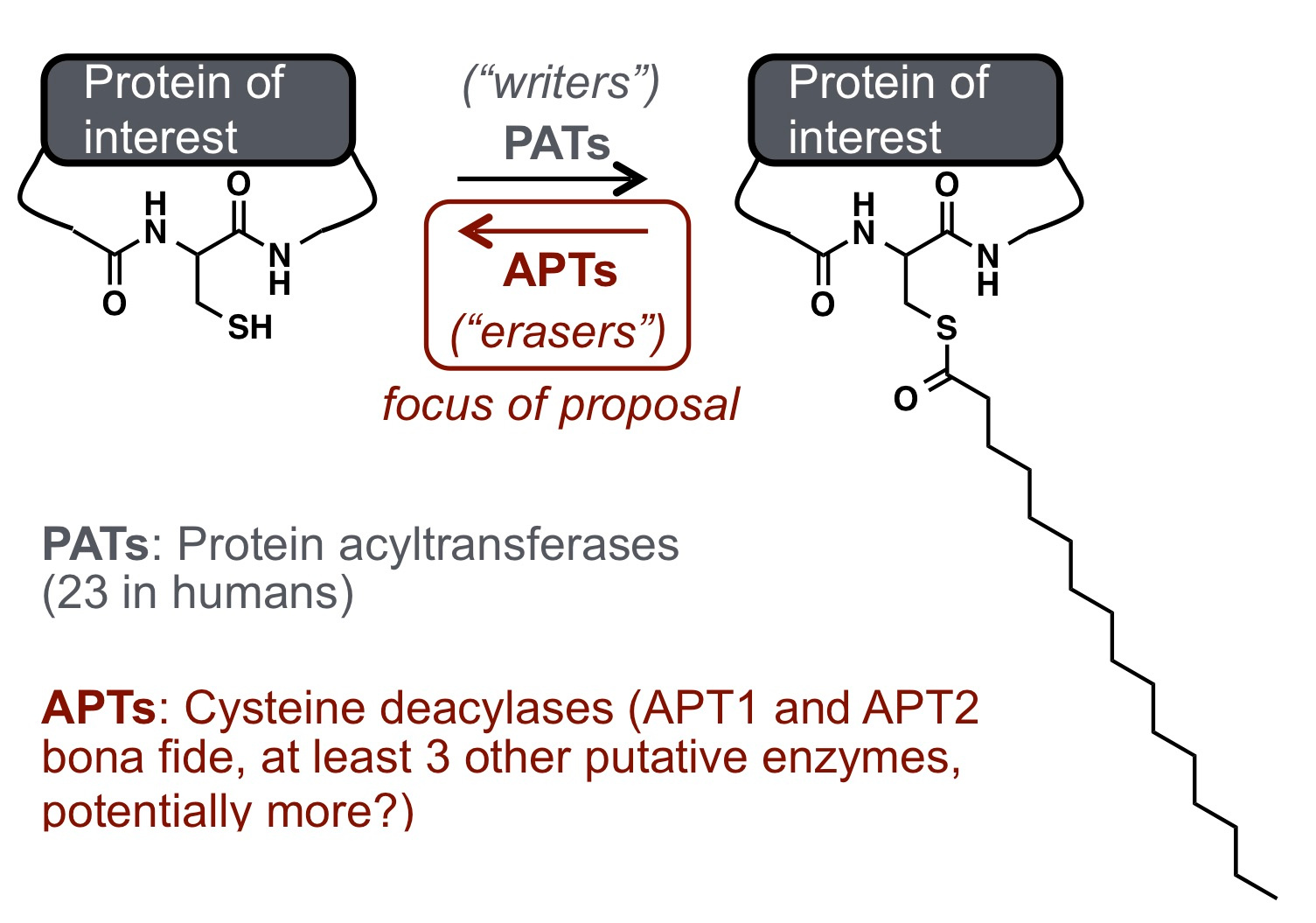
To study the regulation of APTs in live cells, my group has developed a suite of chemical tools including turn-on probes for APTs (https://www.nature.com/articles/nchembio.2262 and https://pubs.acs.org/doi/abs/10.1021/acs.biochem.7b00835) and ratiometric APT probes (https://pubs.rsc.org/en/content/articlehtml/2017/sc/c7sc02805a). We became fascinated with mitochondrial lipidation, because many mitochondrial proteins can be modified by S-acylation-based PTMs, as discovered by a range of unbiased proteomic methods. This was intriguing to us, however, because none of the known APTs were annotated as functional in mitochondria. Therefore, we asked the simple question: is there enzyme-mediated regulation of protein acylation in mitochondria, and if so, who are the players in this regulatory process?
To begin to answer this question, we synthesized a mitochondrial-targeted APT probe, which allowed us to experimentally test whether there are any active enzymes within that compartment. Indeed, using this probe, we discovered that there are APTs in the mitochondria, and to our surprise discovered that APT1, the founding member of this class of regulatory proteins, is partially localized in mitochondria (https://www.nature.com/articles/s41467-017-02655-1). APT1 was annotated as a cytosolic protein over 20 years ago, so our chemical biology approach to assign cellular functions provided critical new insights into the potential role of APTs in mitochondrial signaling. A critical next step, however, was annotating a functional role for APTs in mitochondria.
Our hypothesis was simple: we would use a pan-active APT inhibitor and look for cellular effects connected to mitochondrial function, which would lead us to functions and targets of APT1, since this was the only known mitochondrial APT. As it turns out, this hypothesis was completely wrong, but led us down an intriguing (and incredibly challenging) path. We first discovered mitochondria redox regulation was perturbed by APT inhibitors, and validated it was a mitochondrial-derived effect by developing, validating, and using a new mitochondrial-targeted APT inhibitor. At that point, we thought we only were left with showing APT1 was responsible for the phenotype, at which point we would try to claim this was a mitochondrial APT1-mediated effect. However, APT1 was not the target, as any perturbation of this enzyme showed no effect in our assays.
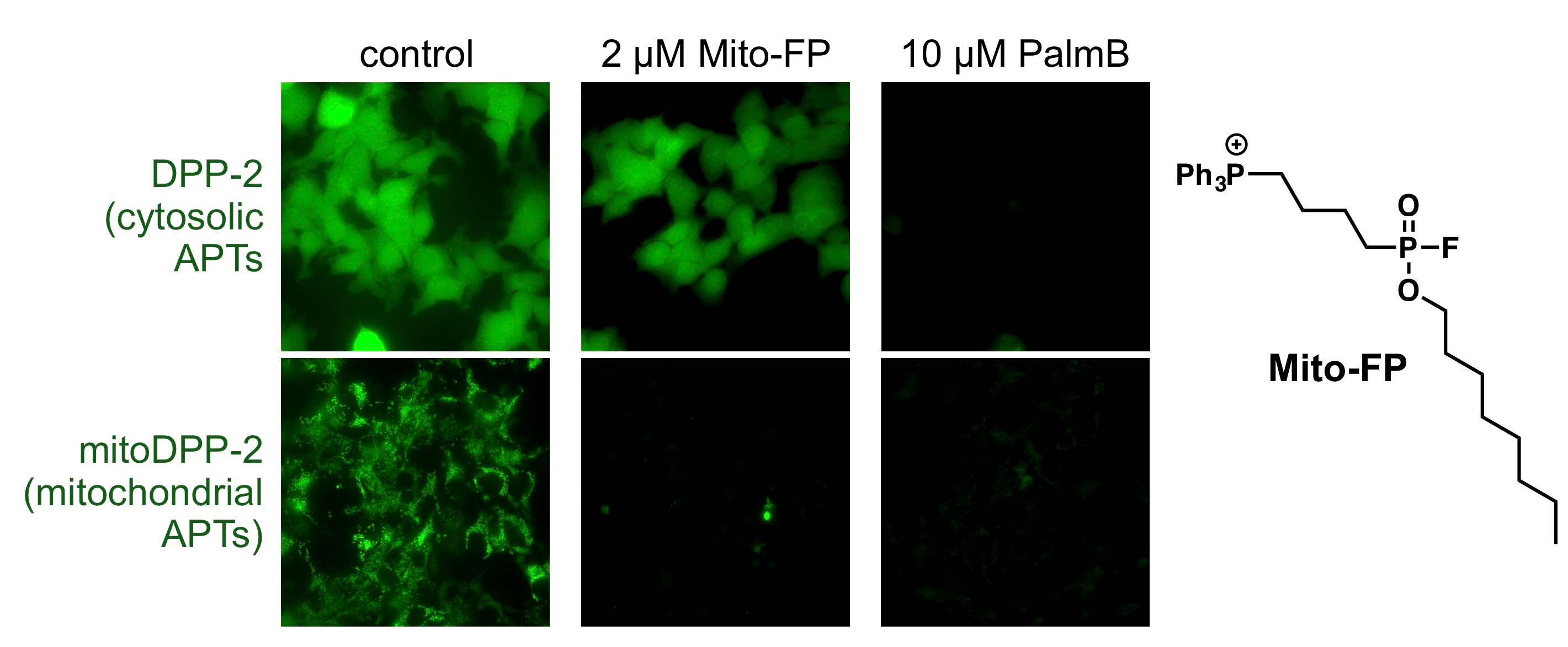
We therefore knew there was a new, unknown enzyme, that we needed to discover. Given the growing complexity of the project, I had team members take different approaches to identify our new enzyme. One group did screening with over-expressed enzymes and our activity probes to identify new proteins with APT activity. A second group did activity-based protein profiling (ABPP) to identify targets of our new inhibitor. To our delight, both of these approaches converged on the same target: ABHD10. This enzyme had no known endogenous function, but was indeed the enzyme we were looking for, as it recapitulated the mitochondrial antioxidant phenotype observed earlier. Through a series of biochemical and cellular studies, we discovered ABHD10 is indeed an APT, and even solved a high-resolution crystal structure of the protein, revealing potential modes of regulation. Finally, we annotated PRDX5 as the first target of ABHD10, and worked out a mechanism by which ABHD10 controls the antioxidant capacity of PRDX5 by direct active site modification.
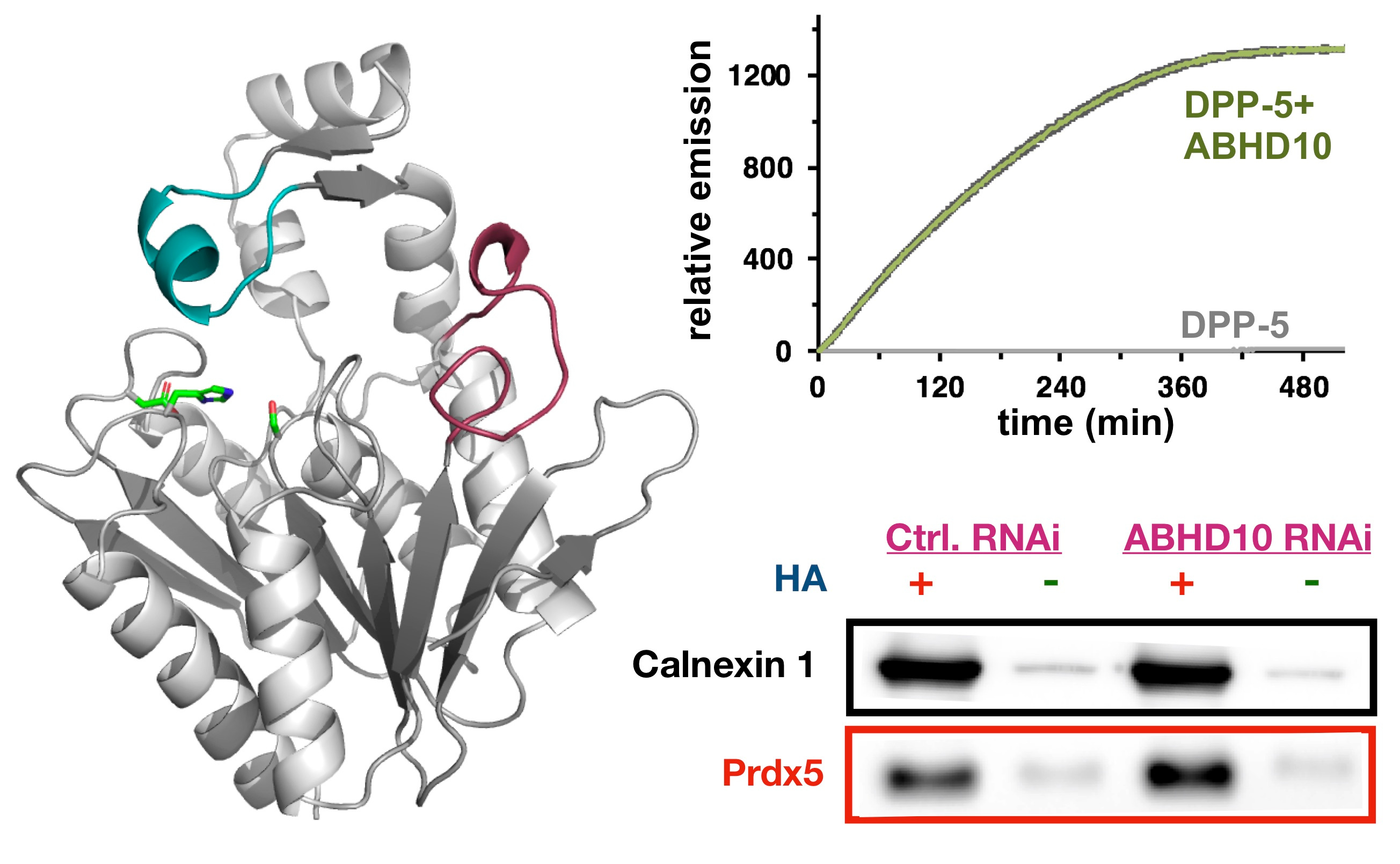
Collectively, this project taught me a few things. First, we can add another enzyme, ABHD10, to the list of APTs, bringing the number of mitochondrial APTs to two. Second, we discovered a new mode of redox regulation by lipidation, which I was certainly not expecting. Third, this work shows that chemical biology-approaches, involving the development of simple probes and crude inhibitors, can really lead to biological discovery. Finally, I learned enzyme annotation is really hard, and the value of team-work to tackle this type of interdisciplinary science. This project required three lead investigators to complete (two graduate students and a post-doc), as well as other collaborators in my group and in other groups. From a managerial perspective, team-based science is never easy. But I truly believe this project would not have come together were it not for the collaborative approach we took.
I really commend Yang, Tian, and Rahul for working together to make this work happen. Saara-Anne, an MD/PhD student in my group, provided critical assistance and help with in vivo experiments. Professor Masaki Fukata and his team helped us with our screen, and my colleague Professor Phoebe Rice helped us with the structure (my group had never done any protein structural biology).
Read more about our work here: https://www.nature.com/articles/s41589-019-0399-y
and a recent review on our technology and discoveries in this area here: https://pubs.acs.org/doi/10.1021/acs.accounts.9b00354
Follow the Topic
-
Nature Chemical Biology
An international monthly journal that provides a high-visibility forum for the chemical biology community, combining the scientific ideas and approaches of chemistry, biology and allied disciplines to understand and manipulate biological systems with molecular precision.





Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in