Circular Vectors better than plasmids for RNA guide delivery and expression in cell culture experiments
Published in Protocols & Methods

We were planning a massive project that aimed to verify which out of tens of thousands of single nucleotide polymorphisms (SNPs) discovered by genome-wide association studies (GWASs) are truly causal. Our project envisioned performing thousands of simple substitution edits to produce cells representing both known states for SNPs and investigating how much gene expression differs nearby the SNPs. Like looking for a bunch of needles in a haystack.
One commonly used approach employs plasmids to deliver and express Cas9 and associated editing machinery into a cell in one large plasmid, which is accompanied by a smaller plasmid expressing an RNA guide targeting a specific location in the genome. These plasmids can be ordered from contract manufacturers. While the expense is not that significant when only a few edits are needed, the cost starts to add up when a larger number of edits is required. This results in many laboratories preparing plasmids themselves via methods that have been used for decades.
A laboratory can prepare massive quantities of the larger Cas9 plasmid in a single 2-day bacterial cloning operation. Such an operation can allow a laboratory to make and purify 10 to 20 (or more) milligrams of this main plasmid, which would be sufficient for a year of experiments. However, the RNA guides targeting specific genomic locations are unique to each edit, whether a simple CRISPR/Cas9, base editing guide RNA, or a more complex guide RNA with a template for prime editing. Only 100–200 nanograms of the smaller plasmid is usually required per editing operation.
The steps involved in preparing such small (typically 2000–3000 base pairs) plasmids expressing the RNA guides are essentially the same as for making the aforementioned large Cas9 plasmid. The workflow includes: a) Designing a specific DNA insert and ordering it as a DNA fragment from a commercial manufacturer; b) Ligating the double-stranded DNA inserts into a selected plasmid vector; c) Using the ligation product in the bacterial transformation, where the tiny amount produced by the ligation of correctly cloned and mis-cloned plasmids is inserted (or “transformed”) into E. coli; d) Plating the transformed E. coli onto agar plates overnight; e) Gene sequencing the colonies, with the ones matching the design being selected and bacteria being grown in a larger volume of liquid culture overnight, followed by the recovery of plasmid DNA using a multistep process; f) Measuring and validating the plasmid concentration via gel electrophoresis, PCR, and sequencing.
Two days later, the plasmid expressing the RNA targeting elements is ready for use in editing operations.
And this is an abridged version of the process; everyone who has done this can list several smaller operations not mentioned here.
We thought, “How nice it would be to skip all the above steps and design a method that would eliminate bacterial cloning altogether? And what if it would cost only a fraction of even in-lab plasmid cloning?” We will not brag. We did not invent the protocol in a feat of inspiration. Instead, we initially tried more complicated approaches. Long story short, it was possible not only to avoid bacterial cloning but to eliminate all the parts of plasmids necessary for the bacterial life cycle. In the form reported in this publication, the Circular Vectors contained a single U6 promoter and a stretch of DNA coding for the intended RNA guide, performed in an optimized by our group single-tube circularization reaction, with reagents costing under $10.
We have succeeded in developing an efficient, fully synthetic method and protocol that enables the production of circularized DNA containing expression elements ready for transfection in as little as 3 hours, thereby eliminating the bacterial cloning steps. In the pictured 3 hour process, we consistently achieved a 48% conversion efficiency of linear DNA into the Circular Vector at lengths of 450 base pairs—a typical length for prime editing guides. The efficiency improves fractionally (up to 62%) with longer ligation duration.
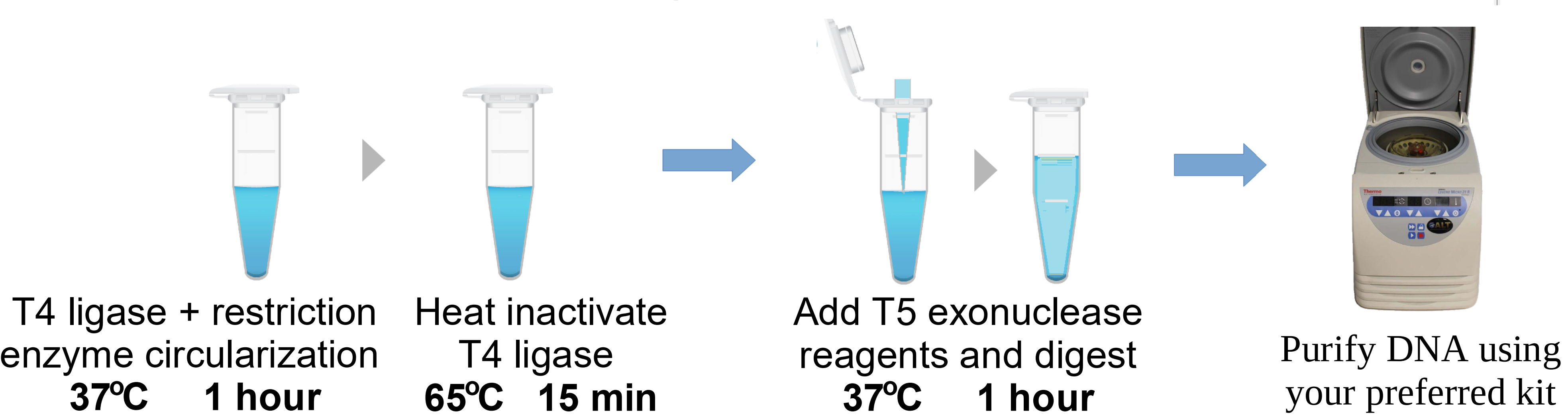
We routinely use this method in our laboratory. Often, we receive linear DNA from a manufacturer in the afternoon and the same evening perform lipofection or nucleofection (two methods used in gene editing) . Even though the reaction efficiency diminishes at lengths over 1000 bp, we have occasionally used our method for ~1500-base pair constructs. We also prepared circularized DNA templates for gene editing via homology-independent targeted integration (HITI), and we had remarkable success in using our Circular Vectors for this purpose. We are certain there are many other applications where our Circular Vector method will prove helpful.
Follow the Topic
-
Communications Biology
An open access journal from Nature Portfolio publishing high-quality research, reviews and commentary in all areas of the biological sciences, representing significant advances and bringing new biological insight to a specialized area of research.

Related Collections
With Collections, you can get published faster and increase your visibility.
Artificial Intelligence Methodology in Structural Biology
Publishing Model: Hybrid
Deadline: Nov 30, 2026
Healthy Aging
Publishing Model: Open Access
Deadline: Dec 31, 2026


Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in