
In the end of 2019, a novel coronavirus disease (COVID-19), caused by a novel type of coronavirus named severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), has been identified in Wuhan, Hubei Province, China. It subsequently spread throughout China and was detected abroad within several weeks, then spark global concern. Super-spreading events (SSEs) described a common phenomenon in many infectious diseases, in which a subset of patients (also called super-spreaders) infects a disproportionate number of secondary cases compared to R0. However, SSEs have not been detected during the early phase of COVID-19 pandemic based on traditional epidemiological tracing approach. It is well known that the epidemiological tracing mainly relies on patients’ recall, which can result in false negatives. Therefore, it is not sure whether the absence of SSEs during the early phase of COVID-19 was the false negatives caused by epidemiological tracing.
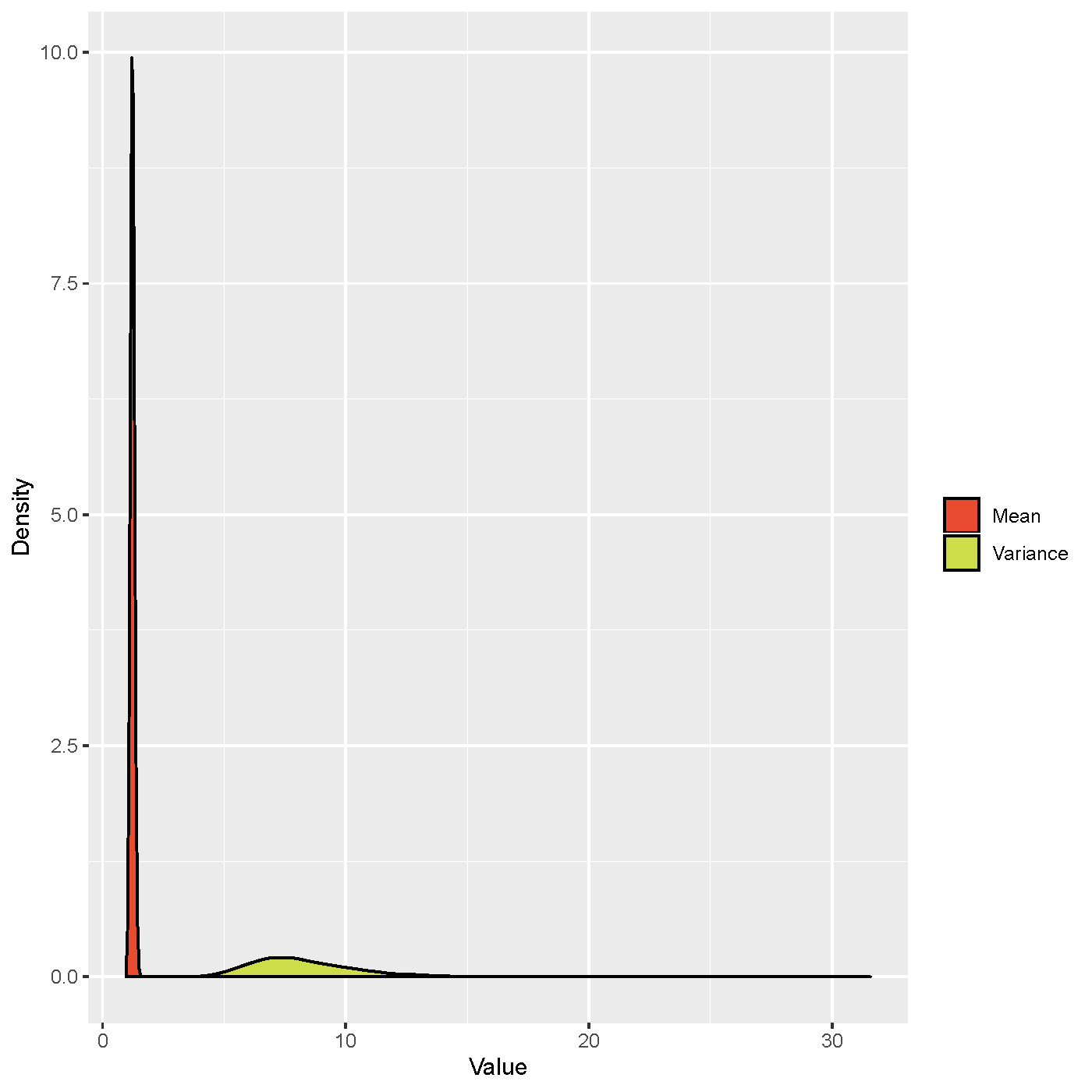
Due to the rapid diagnosis and following sequencing, many SARS-CoV-2 genomes were obtained in a short time. If we could infer SSEs accurately by using viral genomes, it will avoid false negative results caused by epidemiological tracing and also help government to tailor the effective prevention and control strategies to prevent the spread of epidemics. We combined phylogenetic analysis with Bayesian inference under an epidemiological model to trace the person-to-person transmission and estimate the parameters of the offspring distribution during the early outbreak phase of COVID-19. Cross-validation on direct transmission pairs showed that the inferred transmission tree was reliable. Besides, the dispersion parameter of the offspring distribution was estimated to be 0.23 (95% CI: 0.13-0.38), indicating SSEs have occurred early during the outbreak of COVID-19. We also determined that the phylogenetic uncertainty would always overestimate the dispersion parameter of the offspring distribution, indicating underestimation of the extent of SSEs. Our results reveal that it is feasible to use genomic data to identify SSEs.
Follow the Topic
-
Nature Communications
An open access, multidisciplinary journal dedicated to publishing high-quality research in all areas of the biological, health, physical, chemical and Earth sciences.

Related Collections
With Collections, you can get published faster and increase your visibility.
Women's Health
Publishing Model: Hybrid
Deadline: Ongoing
Tumor Microenvironment Crosstalk and Therapeutic Implications
Publishing Model: Hybrid
Deadline: Nov 02, 2026



Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in