Control of human protein-degradation machinery by USP14 revealed in unprecedented detail
Published in Chemistry
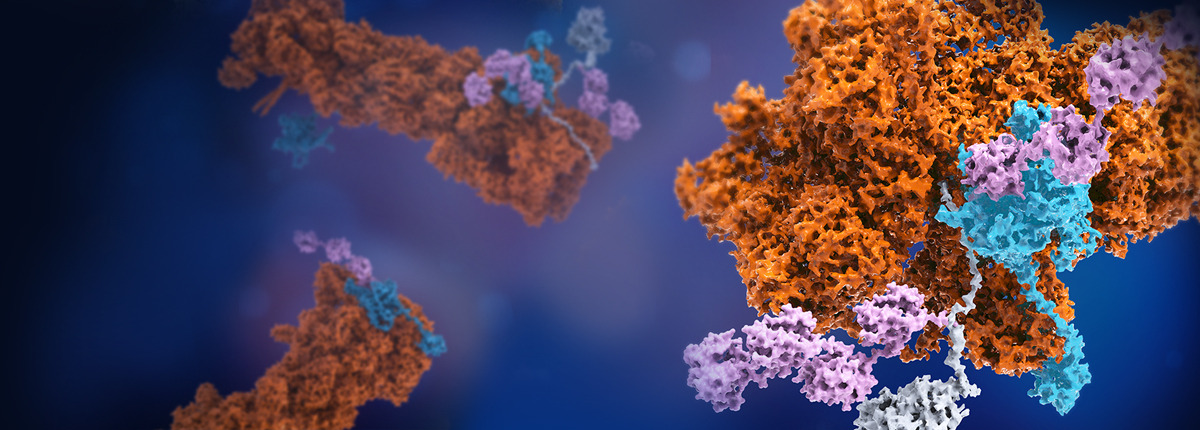

Proteolysis of most of the intracellular proteins in eukaryotes is carried out by the ubiquitin-proteasome system (UPS), which participates in nearly all cellular activities1. The substrate protein to be degraded is first ubiquitylated, a process of attaching ubiquitin molecules, and then targeted for breakdown by a rod-shaped giant proteolytic machinery called the 26S proteasome (Fig. 1a). Once recognizing and capturing the ubiquitylated substrate, the proteasome can mechanically unfold the substrate and translocate it into the proteolytic core named 20S. This chemomechanical process is driven by a ring-like heterohexameric motor module termed AAA-ATPase, which hydrolyses ATP molecules and harnesses the chemical energy to generate mechanical force that unfolds the proteasome-engaged substrate. For many years our group has been studying molecular mechanism of the UPS, especially focusing on the dynamic structures and function of the proteasome. During 2014 to 2018, we have determined atomic structures of the human 26S proteasome using cryo-electron microscopy (cryo-EM) in the absence or presence of ubiquitylated substrates, characterizing the highly dynamic behaviors of the proteasome and its principal working mechanism during substrate degradation2-4.
During our early studies of substrate-engaged proteasomes, we had attempted to capture them right at the moment when the substrates were being unfolded and degraded. However, all these early trials ended up with substantial transient aggregates that defied structural analysis. This trouble was later solved by the trick of replacing ATP with slowly hydrolyzed ATPγS4. This strategy helped to stall many coexisting conformers of the substrate-engaged proteasome in its functional intermediate states4. However, it leaves it an open question if this strategy will introduce certain bias in characterizing the conformational landscape of the proteasome, which may hinder our understanding of how the proteasome is regulated. In the work herein, we managed to take a major step forward and elucidate at a fuller scale the regulation of proteasome function mediated by the deubiquitylating enzyme (DUB) USP14. This ubiquitous DUB has been found to reversibly attach to the proteasome in cells and suppress multiple proteasome activities including degradation of ubiquitylated proteins and RPN11-catalysed deubiquitylation, but contradictorily stimulate certain proteasome activities when bound to polyubiquitin, such as the activity of ATP hydrolysis5. USP14 is able to deubiquitylate a substrate on a millisecond time scale after binding to the proteasome6, which makes it extremely challenging to observe such a transient process at high resolution. To overcome this hurdle, we made various trial and error. We serendipitously found that by adjusting the reaction buffer and lowering the reaction temperature to 10 ℃, both deubiquitylation and proteasomal degradation can be slowed down to such a level that we no longer need to use the ATP-to-ATPγS exchange trick to avoid the unpleasant formation of transient aggregates, which makes it possible for us to observe the USP14-controlled degradation reaction in the presence of ATP in virtually real time. To figure out how USP14 is activated for deubiquitylation and regulates proteasome function, we applied the time-resolved cryo-EM technique, collecting data over several time points after start of the degradation reaction. This eventually enabled atomic-level visualization of structural intermediates of the USP14-proteasome complex and tracking their conformational changes as the proteasome degraded its ubiquitylated substrate.
When we tried to analyze the time-resolved cryo-EM data, a major challenging problem known as heterogeneous reconstruction emerged, namely, how to resolve transient intermediate structures of a protein or complex with sophisticated dynamic behaviors. In nowadays cryo-EM studies, the most common approach is to cluster the data into a few 3D classes using the classification program in Relion7 or cryoSPARC8, which is however not accurate enough to distinguish low-abundance conformers and usually results in mixtures of structures or completely missing transient states, leading to low-resolution reconstructions that defy de novo atomic modeling. With the long-term efforts and experience gained from our structural and methodological studies of macromolecule complexes in the past decade, we have recently developed novel 3D classification methods based on deep learning to improve the accuracy with which we could sufficiently classify different, low-abundance conformational states in a mixture to high resolution9,10. In the final stage, 13 conformers of USP14-proteasome complex in distinct intermediate states have been reconstructed at near-atomic resolution. And the time-dependent changes of the population of the conformational continuum during the proteolysis process were tracked (Fig. 1b).
Our structural findings were further examined by structure-based site-directed mutagenesis assays and revealed how USP14 precisely regulates the proteasome activities in processing its substrate step by step. USP14 is activated for deubiquitylation by binding to the OB ring of the AAA-ATPase motor (Fig. 1a), which hydrolyses ATP around the hexameric ATPase ring and mechanically translocate its substrate. Comparison with previous cryo-EM studies in USP14-free conditions2-4 indicated that the conformational landscape of the AAA-ATPase motor is allosterically reprogrammed upon binding and activation of USP14. A model of complete functional cycle of USP14-proteasome was consequently inferred. Strikingly, USP14 activation creates three checkpoints on the proteasome by inducing parallel pathways of proteasome state transitions, one that inhibits substrate engagement and the other that facilitates substrate degradation.

Figure 1. Time-resolved cryo-EM analysis of the USP14-proteasome complex in the act of substrate degradation. (a) Cryo-EM structure of the substrate-engaged USP14-proteasome complex in state ED2.1USP14, one of the 13 intermediate states. USP14 in this state is bound to the AAA-ATPase ring. (b) Kinetic changes of the particle populations of 13 conformational states of the USP14-bound proteasome over time after addition of the substrate.
The proteasome function has to be tightly regulated at many levels. This makes it an outstanding problem of extreme complication, considering of many proteasome-regulating proteins with unknown mechanisms and diverse upstream ubiquitin signals that determine the substrate fate1,5,11. Despite considerable knowledge of USP14 regulation of the proteasome described in this paper, many other important cofactors, such as substrate-shuttling proteins, or enzymes of ubiquitination, can also associate with the proteasome and are implicated in various human diseases1. So, it will be fascinating and important to study how these proteins modulate proteasome function in the future.
Solving conformational continuum or structural ensemble at high resolution in the course of actual biochemical reaction has been largely prohibited in the past. The current study presents an emerging paradigm in elucidating the molecular mechanism of enzymatic actions by reconstituting functional kinetics of the reaction system in vitro and then imaging the entire process directly down to the atomic level. Such an approach appears to afford less inference by reconstructing high-resolution intermediate states that intuitively inform molecular mechanisms. Thus, we optimistically envision that such a paradigm may help shape up the future biochemistry and molecular biology as well as find key applications in therapeutic discovery.
For more details on this work, please read our paper in Nature at https://doi.org/10.1038/s41586-022-04671-8.
Related Nature Research Briefing at https://doi.org/10.1038/d41586-022-01144-w.
Written By Youdong Mao & Shuwen Zhang at Peking University
References
1 Mao, Y. Structure, Dynamics and Function of the 26S Proteasome. Subcell Biochem 96, 1-151, https://doi.org/10.1007/978-3-030-58971-4_1 (2021).
2 Chen, S. et al. Structural basis for dynamic regulation of the human 26S proteasome. Proc Natl Acad Sci U S A 113, 12991-12996, https://doi.org/10.1073/pnas.1614614113 (2016).
3 Zhu, Y. et al. Structural mechanism for nucleotide-driven remodeling of the AAA-ATPase unfoldase in the activated human 26S proteasome. Nat Commun 9, 1360, https://doi.org/10.1038/s41467-018-03785-w (2018).
4 Dong, Y. et al. Cryo-EM structures and dynamics of substrate-engaged human 26S proteasome. Nature 565, 49-55, https://doi.org/10.1038/s41586-018-0736-4 (2019).
5 de Poot, S. A. H., Tian, G. & Finley, D. Meddling with Fate: The Proteasomal Deubiquitinating Enzymes. J Mol Biol 429, 3525-3545, https://doi.org/10.1016/j.jmb.2017.09.015 (2017).
6 Lee, B. H. et al. USP14 deubiquitinates proteasome-bound substrates that are ubiquitinated at multiple sites. Nature 532, 398-401, https://doi.org/10.1038/nature17433 (2016).
7 Zivanov, J. et al. New tools for automated high-resolution cryo-EM structure determination in RELION-3. Elife 7, https://doi.org/10.7554/eLife.42166 (2018).
8 Punjani, A., Rubinstein, J. L., Fleet, D. J. & Brubaker, M. A. cryoSPARC: algorithms for rapid unsupervised cryo-EM structure determination. Nat Methods 14, 290-296, https://doi.org/10.1038/nmeth.4169 (2017).
9 Wu, Z. et al. Hidden dynamics of proteasome autoregulation discovered by cryo-EM data-driven deep learning. bioRxiv, https://doi.org/10.1101/2020.12.22.423932 (2022).
10 Wu, Z. et al. Visualizing conformational space of functional biomolecular complexes by deep manifold learning. Int J Mol Sci 23, 8872, https://doi.org/10.3390/ijms23168872 (2022).
11 Komander, D. & Rape, M. The ubiquitin code. Annu Rev Biochem 81, 203-229, https://doi.org/10.1146/annurev-biochem-060310-170328 (2012).
Follow the Topic
-
Nature
A weekly international journal publishing the finest peer-reviewed research in all fields of science and technology on the basis of its originality, importance, interdisciplinary interest, timeliness, accessibility, elegance and surprising conclusions.

Related Collections
With Collections, you can get published faster and increase your visibility.
Carbon Dioxide Removal
Publishing Model: Hybrid
Deadline: Jan 16, 2027

Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in