Core and accessory genomic traits of Vibrio cholerae O1 drive lineage transmission and disease severity
Published in Microbiology, Protocols & Methods, and Mathematics

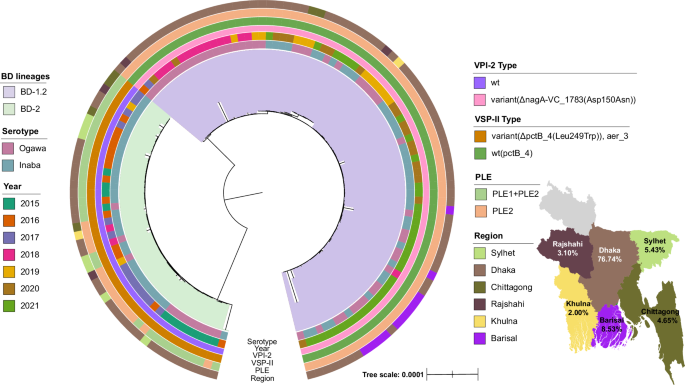
Cholera is a severe diarrheal illness that affects millions worldwide. It is estimated that around 1.3 billion people are at risk globally, with annual case numbers ranging from 1.3 to 4 million1,2, leading to 21,000 to 143,000 deaths1,3. In Bangladesh, where cholera is endemic, approximately 66 million people are at risk, with the country experiencing around 100,000 cases and 4,500 deaths each year. About 1 in 5 people with cholera will experience a severe condition owing to a combination of symptoms (primarily diarrhoea, vomiting, dehydration)4. Furthermore, Vibrio cholerae demonstrates a remarkable ability to evolve genetically and phenotypically, resulting in successive pandemic clones with varying degrees of virulence, pathogenicity, resistance, and spreading capability5,6.
Distinctive lineages belonging to the 7th pandemic El Tor wave-3 have been observed circulating in Bangladesh7-9. Recent studies have identified key mutations in lineages such as BD-1 and BD-27-9 and the emergent BD-1.28, but gaps remain in fully understanding the genomic factors behind transmission dynamics and disease severity. In this study, we analysed in-patients isolates from six Bangladeshi regions between 2015 and 2021. Two main objectives were set: 1. identify the genetic variations and signatures of the BD-1.2 lineage evolution beyond what has been found so far; 2. investigate if correlations exist between the genomic determinants of BD-1.2 strains and clinical manifestations among hospitalised patients from whom the isolates were collected from. Therefore, to achieve them, we developed a novel computational framework combining machine learning (ML), genome-scale metabolic modelling (GSMM), and 3D structural analysis.
Our approach first identified a broader set of genomic determinants (accessory genes, coding and non-coding SNPs) separating the lineages BD-1.2 and BD-2, which expand upon previous findings8. Key mutations in genes related to toxin transport, acid tolerance, virulence, growth, response to pH and temperature, biofilm formation, and bacteriophage resistance were discovered, providing insights into the dominance of BD-1.2 over BD-2. From the annotated accessory genes that statistically separate the two lineages, five (lon_3, endA, adh, hdfR_4 and bcr_2) were present uniquely in BD-1.2 with functions such as antibiotic resistance and biofilm formation. Moreover, the GSMM analysis identified genes playing important metabolic roles.
Roughly 20% of people who contract V. cholerae show moderate to severe cholera symptoms4. Here, we revealed the existence of correlations between a core set of genetic determinants in V. cholerae and clinical symptoms (diarrhoeal duration, number of stools, abdominal pain, vomit, and dehydration) using a machine learning framework. Overall, the analysis revealed an overlap of genetic determinants associated with BD-1.2 transmission and to the clinical symptoms linked to this lineage. Additionally, a distinct set of genetic determinants were found exclusively linked to the severity of clinical symptoms. Furthermore, our GSMM analysis indicated that some of these genes when knocked out were predicted to have important bacterial metabolic functions. Our 3D structural analysis also showed that mutations in genes present in BD-1.2 (important for both the lineage separation analysis and the correlation with clinical symptoms) increased protein stability, potentially contributing to more severe disease.
In conclusion, our study provides a comprehensive view of the genetic determinants driving V. cholerae lineage evolution and its link to clinical severity. The integration of ML, GSMM, and structural analysis has enabled the identification of novel associations and key genomic traits, offering insights into cholera transmission and pathogenesis. These findings could guide future therapeutic strategies and interventions to mitigate the spread and impact of cholera.
References
1 Baddam, R. et al. Genome dynamics of Vibrio cholerae isolates linked to seasonal outbreaks of cholera in Dhaka, Bangladesh. MBio 11, e03339-03319 (2020).
2 Ali, M., Nelson, A. R., Lopez, A. L. & Sack, D. A. Updated Global Burden of Cholera in Endemic Countries. PLoS Negl. Trop. Dis. 9, e0003832, doi:10.1371/journal.pntd.0003832 (2015).
3 Banerjee, R., Das, B., Nair, G. B. & Basak, S. Dynamics in genome evolution of Vibrio cholerae. Infect. Genet. Evol. 23, 32-41 (2014).
4 Baker-Austin, C. et al. Vibrio spp. infections. Nat Rev Dis Primers 4, 8, doi:10.1038/s41572-018-0005-8 (2018).
5 Wozniak, R. A. et al. Comparative ICE genomics: insights into the evolution of the SXT/R391 family of ICEs. PLoS Genet. 5, e1000786, doi:10.1371/journal.pgen.1000786 (2009).
6 Faruque, S. M. & Mekalanos, J. J. Pathogenicity islands and phages in <em>Vibrio cholerae</em> evolution. Trends Microbiol. 11, 505-510, doi:10.1016/j.tim.2003.09.003 (2003).
7 Monir, M. M. et al. Genomic Characteristics of Recently Recognized Vibrio cholerae El Tor Lineages Associated with Cholera in Bangladesh, 1991 to 2017. Microbiology Spectrum 10, e00391-00322 (2022).
8 Monir, M. M. et al. Genomic attributes of Vibrio cholerae O1 responsible for 2022 massive cholera outbreak in Bangladesh. Nat. Commun. 14, 1154, doi:10.1038/s41467-023-36687-7 (2023).
9 Morita, D. et al. Whole-genome analysis of clinical Vibrio cholerae O1 in Kolkata, India, and Dhaka, Bangladesh, reveals two lineages of circulating strains, indicating variation in genomic attributes. Mbio 11, e01227-01220 (2020).
Follow the Topic
-
Nature Communications
An open access, multidisciplinary journal dedicated to publishing high-quality research in all areas of the biological, health, physical, chemical and Earth sciences.

Related Collections
With Collections, you can get published faster and increase your visibility.
Women's Health
Publishing Model: Hybrid
Deadline: Ongoing
Tumor Microenvironment Crosstalk and Therapeutic Implications
Publishing Model: Hybrid
Deadline: Nov 02, 2026

Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in