Coverage-driven selectivity switch from ethylene to acetate in high-rate CO2/CO electrolysis
Published in Chemistry


As an emerging carbon capture, utilization, and storage technology, CO2 electrolysis to valuable chemicals and fuels with power supply derived from renewable energy is of great importance1. To push the CO2 electrolysis process towards practical application, achieving high selectivity of multicarbon (C2+) products (e.g., ethylene, acetate, ethanol, and n-propanol) at industrial current densities is highly desirable2. Apart from rational design of catalytically active nanostructures, tuning the microenvironments near catalyst surfaces has also been demonstrated to be effective in facilitating C–C coupling and improving C2+ production. Catalyst microenvironments can be modified via tuning electrolyte compositions (e.g., cation, anion, and pH) as well as introducing organic compounds and ionomer layers on catalyst surfaces3–7. A key role of these tailored microenvironments is to vary the local concentrations of CO2, H2O, OH− and H+ as well as the surface coverages of key intermediates such as adsorbed CO (*CO). An alternative way to modify catalyst microenvironments is to use mixed CO/CO2 feeds that are usually derived from incomplete industrial combustion of fossil fuels. Yet, the microenvironment in the presence of mixed CO/CO2 feeds and its effects on the formation of C2+ products are poorly understood. Currently, most of mechanistic studies focus on pure CO2 and CO electrolysis, and only a few of them take account of CO-CO2 co-electrolysis8,9. For example, a recent study proposed a CO/CO2 cross-coupling mechanism that effectively explains improved ethylene production in an H-cell with a neutral electrolyte9. A membrane electrode assembly (MEA) electrolyzer is considered as the most promising electrochemical device for CO2 electrolysis, however, to date fundamental understandings on the reaction mechanism of CO/CO2 co-electrolysis under alkaline MEA conditions are still lacking.
In the present work, we investigated CO/CO2 co-electrolysis over nanoporous CuO nanosheets in an alkaline MEA electrolyzer to fill the knowledge gap of both electrocatalytic reactivity and mechanistic understanding. In the pure CO2 feed, CO2 electrolysis delivered a peak ethylene Faradaic efficiency (FE) of 52.5% at an applied current density of 0.6 A cm−2. When CO/CO2 feeds were fed to the electrolyzer, the product selectivity shifted from ethylene to acetate. With increasing CO pressure in the feed, acetate replaced ethylene as the major product and meanwhile the total current density significantly increased. In 0.6 MPa CO feed, the acetate FE achieved 48% with a total current density of 3 A cm−2. Under optimized conditions, the FE and partial current density of C2+ products reached 90.0% and 3.1 A cm−2, corresponding to a carbon selectivity of 100.0% and yield of 75.0%, outperforming thermocatalytic CO hydrogenation (Figure 1).
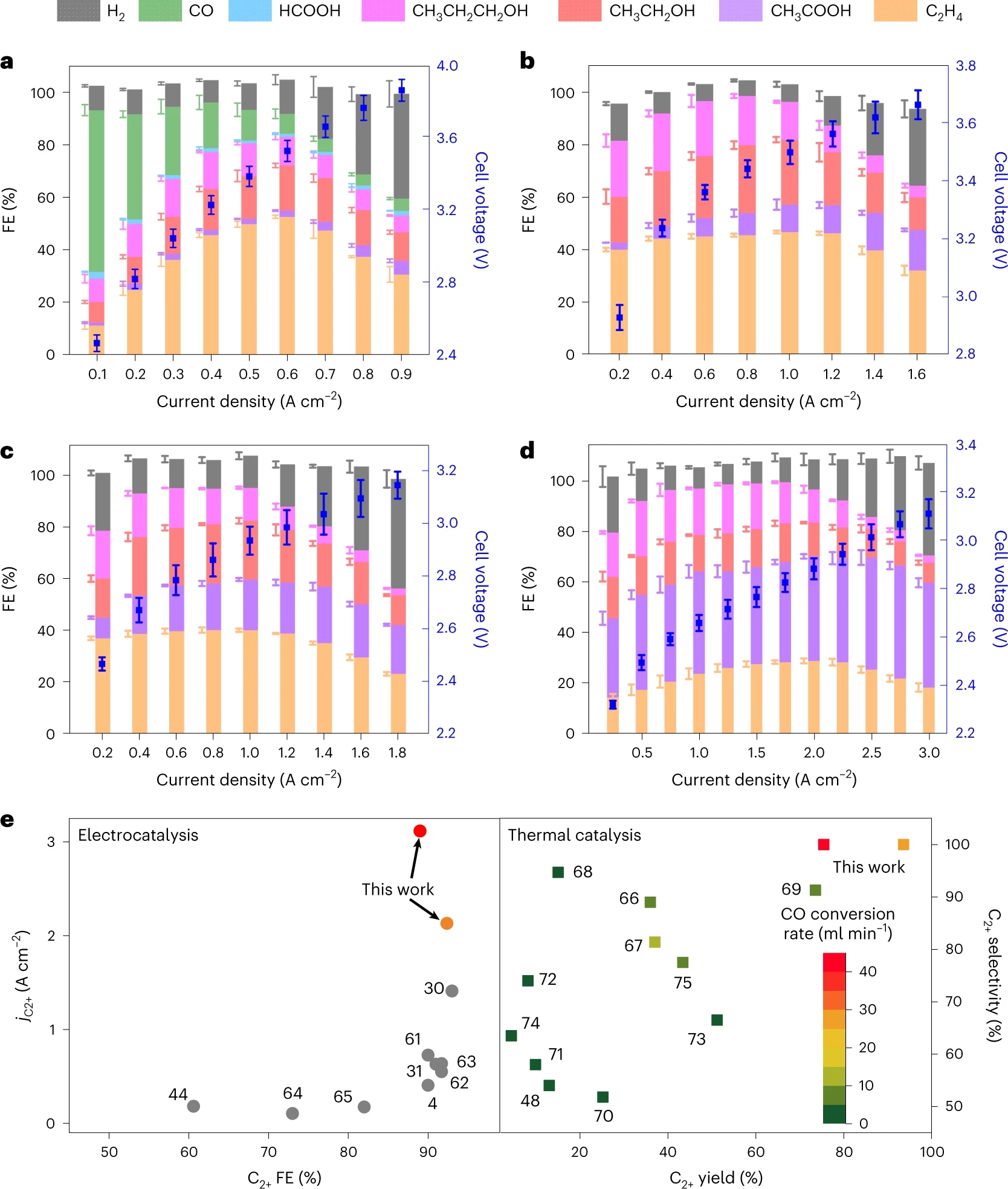
Figure 1. CO2/CO electrolysis performance. a–d, FE and cell voltage as a function of applied current density over CuO nanosheet catalyst measured in 0.1 M KOH under pure CO2 feed (a), CO/CO2 (3:1) co-feed (b), pure CO feed (c) and 1 M KOH under 0.6 MPa pure CO feed (d). e, Comparison of CO reduction via electrocatalysis and thermal catalysis. CO flow rates of 30 ml min−1 (red) and 60 ml min−1 (orange).
To figure out the selectivity switch from ethylene to acetate, we firstly characterized the CuO nanosheet catalyst in its as-prepared state, during and after CO2/CO electrolysis. While the CuO nanosheet catalyst suffered from significant reconstruction, the changes in the morphology and structure were independent on feed composition. The 13CO isotopic labelling experiment followed by GC-MS product analysis indicated that only 2.4% of the CO2 in the CO/CO2 (3:1) co-feed went to products and the CO/CO2 cross-coupling mechanism could not explain the improved ethylene production under alkaline MEA conditions. Decreasing CO partial pressure and local pH are two roles played by CO2 in the co-feeds, and their effects were decoupled by replacing CO2 with Ar and pressurizing CO. The higher local pH did play a role in acetate production, however, the selectivity switch from ethylene to acetate was mainly driven by CO pressure (*CO coverage).
Operando Raman spectroscopy measurements were conducted to observe surface adsorbate species during electrolysis. The product distribution measured in the operando cell almost resembled that measured in our MEA electrolyzer. We found that with increasing CO pressure the atop-adsorbed *CO band shifted towards higher wavenumber and the area ratio of high-frequency-band (HFB)-*COatop and low-frequency-band (LFB)-*COatop increased from 0.73 in the CO/CO2 (1:1) co-feed to 2.74 in 0.4 MPa pure CO feed. In combination with literature10, we postulate that CO tends to preferentially adsorb on terrace sites at low *CO coverage and step sites are provided with more CO at high *CO coverage. Density functional theory (DFT) calculations further indicate that the ethylene formation is favorable on Cu(100) facet (terrace sites) while the acetate formation is significantly improved on Cu(511) facet (step sites). The identification of reaction sites by operando Raman spectroscopy and the analysis of reaction pathways after C−C coupling step by DFT calculations rationalize the *CO coverage-driven selectivity switch from ethylene to acetate in high-rate CO2/CO electrolysis (Figure 2).
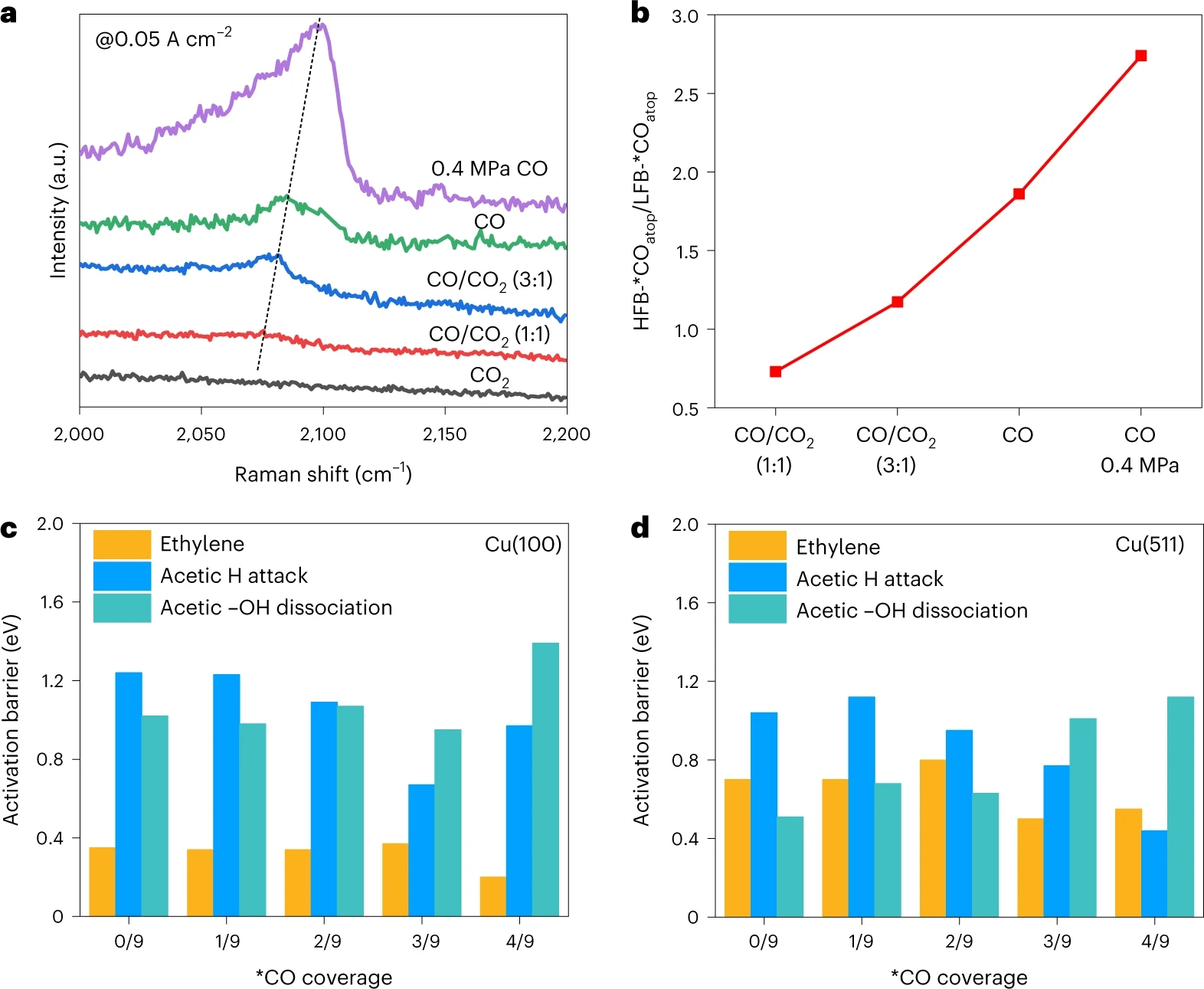
Figure 2. *CO-coverage-dependent reaction pathways. a, Operando *COatop Raman peaks of the CuO nanosheet catalyst measured in 0.1 M KOH at 0.05 A cm−2. b, Peak area ratio of HFB-*COatop to LFB-*COatop. c,d, Activation free-energy barrier of ethylene as well as acetate via H attack and −OH dissociation on Cu(100) (c) and Cu(511) (d).
To push CO2/CO electrolysis towards practical application, the scale-up of the electrolysis is indispensable. We firstly scaled up the electrolysis process by increasing the geometric electrode area from 4 to 100 cm2 and then assembled an electrolyzer stack with four 100 cm2 MEAs. For CO2 electrolysis, a peak ethylene FE of 49.9% was achieved at a total current of 40 A. For CO electrolysis, the C2+ FE was still higher than 65% at a total current of 250 A, with the highest ethylene formation rate of 457.5 mL min–1 at 150 A and acetate formation rate of 2.97 g min–1 at 250 A. The electrolysis performances are comparable in the 4 and 100 cm2 electrolyzers as well as 100 cm2 stack, highlighting the excellent effectiveness of our scale-up demonstration (Figure 3).
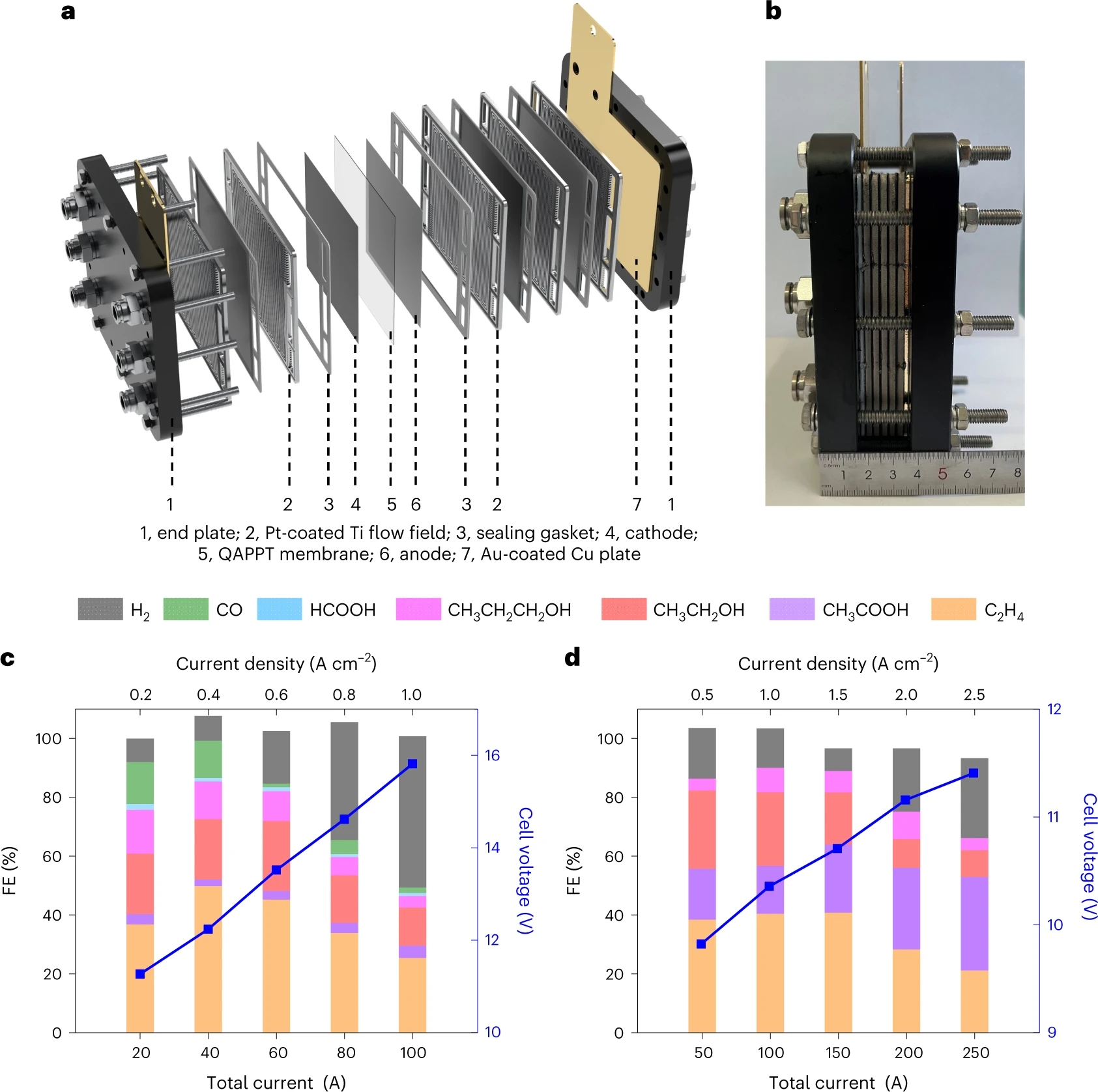
Figure 3. Scale-up demonstration. a,b, Schematic (a) and photograph (b) of the electrolyser stack with four 100 cm2 MEAs used in this work. c,d, FE and cell voltage as a function of applied current density over CuO nanosheet catalyst measured in 0.1 M KOH under pure CO2 feed (c) and 1 M KOH under pure CO feed (d).
Our work highlights the promise of tuning catalyst microenvironments for the selective production of single C2+ products such as acetate and ethylene, and presents an effective scale-up demonstration of high-rate CO2/CO electrolysis.
If you would like to know more details, please take a look at our article published in Nature Nanotechnology: https://doi.org/10.1038/s41565-022-01286-y
References
- Bushuyev, O. S. et al. What should we make with CO2 and how can we make it? Joule 2, 825–832 (2018).
- Masel, R. I. et al. An industrial perspective on catalysts for low-temperature CO2 Nat. Nanotechnol. 16, 118–128 (2021).
- Kim, C. et al. Tailored catalyst microenvironments for CO2 electroreduction to multicarbon products on copper using bilayer ionomer coatings. Energy 6, 1026–1034 (2021).
- Yan, Z. et al. Improving the efficiency of CO2 electrolysis by using a bipolar membrane with a weak-acid cation exchange layer. Chem. 13, 33–40 (2021).
- Monteiro, M. C. O. et al. Absence of CO2 electroreduction on copper, gold and silver electrodes without metal cations in solution. Catal. 4, 654–662 (2021).
- Kim, D. et al. Selective CO2 electrocatalysis at the pseudocapacitive nanoparticle/ordered-ligand interlayer. Energy 5, 1032–1042 (2020).
- Huang, J. et al. CO2 electrolysis to multicarbon products in strong acid. Science 372, 1074–1078 (2021).
- Lum, Y. & Ager, J. W. Evidence for product-specific active sites on oxide-derived Cu catalysts for electrochemical CO2 Nat. Catal. 2, 86–93 (2019).
- Wang, X. et al. Mechanistic reaction pathways of enhanced ethylene yields during electroreduction of CO2-CO co-feeds on Cu and Cu-tandem electrocatalysts. Nanotechnol. 14, 1063–1070 (2019).
- Gunathunge, C. M. et al. Revealing the predominant surface facets of rough Cu electrodes under electrochemical conditions. ACS Catal. 10, 6908–6923 (2020).
Follow the Topic
-
Nature Nanotechnology
An interdisciplinary journal that publishes papers of the highest quality and significance in all areas of nanoscience and nanotechnology.





Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in