Cracking the Code of Pancreatic Cancer Spread: A New Role for ALK7
Published in Bioengineering & Biotechnology, Materials, and Biomedical Research

Pancreatic ductal adenocarcinoma (PDAC) is one of the deadliest cancers known today. Despite years of intensive research, the survival statistics remain devastating: fewer than one in ten patients live beyond five years after diagnosis, and the majority die from metastatic disease within just two years. What makes PDAC especially lethal is not just its aggressive biology, but also its unique tumor microenvironment—a dense, fibrous stroma that surrounds the tumor like armor. This fibrotic shield makes drug delivery difficult and physically restrains the movement of tumor cells, yet paradoxically, PDAC cells still manage to spread with remarkable efficiency.
Unlike many other cancers, where tumor cells readily exit blood vessels (extravasation) to colonize distant organs, PDAC faces a bigger challenge earlier in the process: entering the bloodstream in the first place. This step, called intravasation, is a critical rate-limiting barrier. Tumor cells must squeeze past the fibrotic stroma, degrade the surrounding extracellular matrix (ECM), and breach the vascular wall before they can circulate. Once inside, the rest of the journey is comparatively less restrictive. Understanding exactly how PDAC cells achieve this feat has been a long-standing puzzle—and solving it could open new therapeutic doors.
The Missing Piece: ALK7
Much of cancer biology has centered on the transforming growth factor beta (TGFβ) family of receptors, known to regulate cell growth, survival, and metastasis. Among them, activin receptor-like kinase 7 (ALK7) has been something of a mystery. In some cancers, ALK7 appeared to suppress metastasis, while in others, it seemed to fuel it. These contradictory findings have slowed the development of ALK7-targeted therapies, leaving its true role unresolved.
In PDAC, the question has been especially pressing. Advanced pancreatic tumors often exploit TGFβ signaling to resist anoikis—a form of programmed cell death triggered when cells detach from the ECM. By hijacking this pathway, cancer cells survive in circulation and metastasize more efficiently. But how ALK7 fits into this landscape remained unclear.
Two Roads to Spread: The Dual Role of ALK7
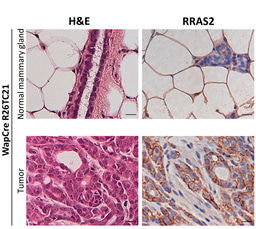
Our new study sheds new light on this mystery by uncovering two interconnected ALK7-driven pathways that empower PDAC cells to metastasize.
-
ALK7–β-catenin–EMT axis:
This pathway boosts the intrinsic mobility of tumor cells. Through activation of epithelial-mesenchymal transition (EMT), cells loosen their adhesive ties and adopt a more flexible, motile identity. EMT is a well-established driver of metastasis, allowing cells to slip past barriers that would otherwise confine them. -
ALK7–β-catenin–MMP axis:
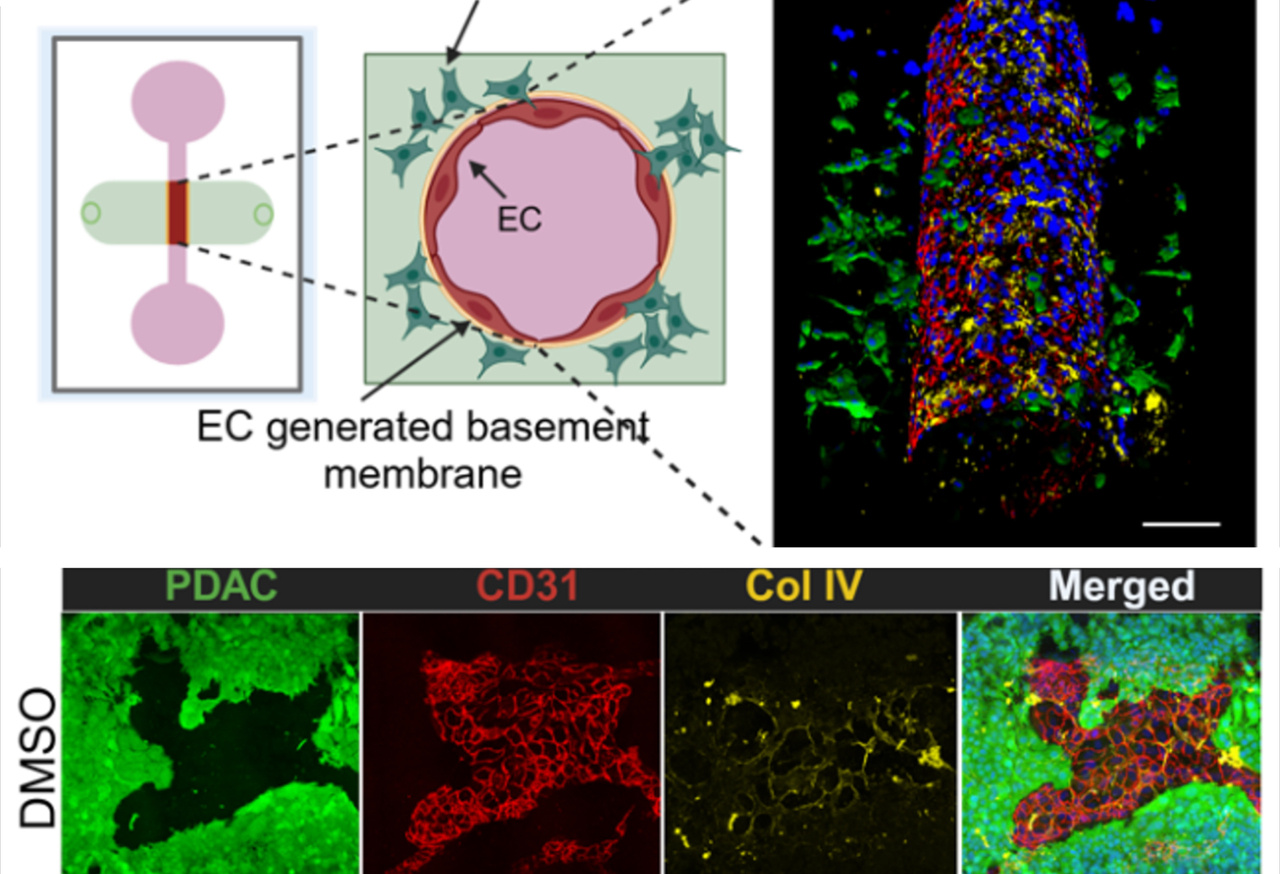
In parallel, ALK7 drives the production of matrix metalloproteinases (MMPs)—enzymes that chew through the ECM and basement membrane. This leads to the formation of invadosomes, specialized cellular structures that physically break down vascular barriers. Also, it breaks down the vascular basement membrane, making the vessels more permeable. The result? Tumor cells gain the ability to intravasate more efficiently, entering the bloodstream and setting the stage for distant spread.
Together, these pathways provide PDAC cells with both the “engine” (motility) and the “tools” (ECM degradation) to invade blood vessels.
Proof in Models: Blocking ALK7 Slows Spread
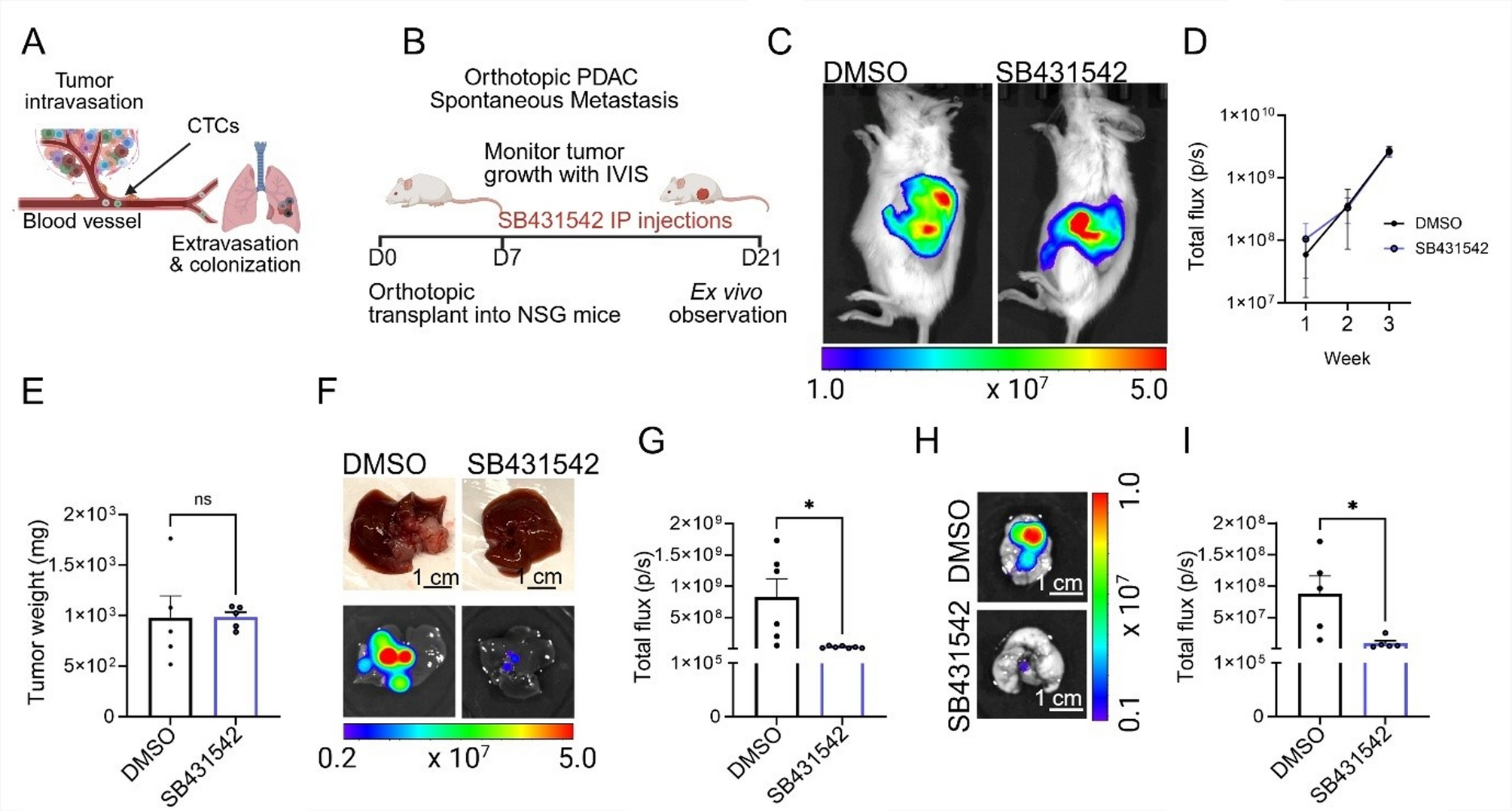
The study combined orthotopic PDAC models (tumors implanted in their natural pancreatic environment in mice) with advanced 3D microfluidic vessel-on-chip platforms that mimic human blood vessels. Both systems pointed to the same conclusion: inhibiting ALK7 slows metastasis.
-
Pharmacological and genetic ALK7 inhibition suppressed the spread of pancreatic tumors in mice.
-
In engineered vessel-on-chip systems, blocking ALK7 preserved the integrity of the basement membrane and limited tumor cell intravasation.
Interestingly, while MMP inhibition effectively blocked vascular barrier breakdown and intravasation, it did not affect extravasation—the later stage where cells exit blood vessels into new tissues. This distinction underscores a key finding: different molecular pathways govern different stages of metastasis. For PDAC, intravasation is the bottleneck, and ALK7 is a central player.
Why This Matters
These discoveries position ALK7 as a dual-function pro-metastatic regulator in PDAC. By orchestrating both tumor cell plasticity and ECM remodeling, ALK7 equips cancer cells with the means to break through the most formidable barrier they face: the vascular wall.
For patients, this is more than just a mechanistic insight. It points toward a promising therapeutic opportunity. If drugs can be designed or repurposed to inhibit ALK7, they could disrupt PDAC metastasis at its earliest and most vulnerable stage—before cancer cells enter circulation.
Looking Ahead
PDAC has long been considered one of the most intractable cancers, in large part because by the time it is diagnosed, metastasis is often already underway. The identification of ALK7 as a key driver of intravasation opens new avenues for intervention. Future work will need to refine ALK7 inhibitors, test their efficacy in combination with existing chemotherapies, and assess their safety in clinical settings.
If successful, this strategy could finally tip the scales in favor of patients, slowing the relentless spread of pancreatic cancer and improving survival.
In a disease where time is often tragically short, targeting ALK7 could offer patients something precious: a chance at more time.
Lee Lab develops and engineers the three-dimensional (3D) microphysiological systems (MPS) of human lymphatics and vasculatures to provide new therapeutic strategies for lymphatic and vasculature-related diseases, including lymphedema, inflammatory disease, cancer, and neurological (brain and eye) disease.
Follow the Topic
-
Molecular Cancer
This journal promotes the exchange of ideas, concepts and findings in any area of cancer and related biomedical science, from a molecular point of view.

Related Collections
With Collections, you can get published faster and increase your visibility.
3D Cancer Models: Organoids and Advanced Culture Systems in Therapy and Drug Resistance
This Collection focuses on the latest advances in 3D culture technologies, including organoids, spheroids, and biomimetic systems, as transformative tools for human cancer research. These models better replicate tumor architecture, cell-cell and cell-matrix interactions, and therapeutic response, bridging the gap between in vitro assays and in vivo complexity. We welcome original research and reviews that explore the use of 3D systems to study cancer progression, drug resistance, immunotherapy, and precision medicine applications.
All submissions in this collection undergo the journal’s standard peer review process. Similarly, all manuscripts authored by a Guest Editor(s) will be handled by the Editor-in-Chief. As an open access publication, this journal levies an article processing fee (details here). We recognize that many key stakeholders may not have access to such resources and are committed to supporting participation in this issue wherever resources are a barrier. For more information about what support may be available, please visit OA funding and support, or email OAfundingpolicy@springernature.com or the Editor-in-Chief.
Publishing Model: Open Access
Deadline: Jun 01, 2026
Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in